本 期 要 目
[监管动态]
全国药品监管工作座谈会强调 持续深化改革 奋力谱写新时代药监工作新篇章
国家药品监督管理局发布《接受药品境外临床试验数据的技术指导原则》
国家药品监督管理局修订清开灵注射剂和注射用益气复脉(冻干)等3个说明书
国家药品监督管理局发布31批次药品不符合规定的通告
国家药品监督管理局发布角膜塑形用硬性透气接触镜及软性接触镜2项临床试验指导原则
[综合分析]
2018年6月CDE药品审评情况分析报告
年中盘点,2018年国内批准了这些药品
[行业信息]
从审批到监管 中国医疗器械法规体系有什么变化
未来十年医疗器械行业发展前景
[国外信息]
无菌企业自检清单——FDA检查员手册的168个问题
美国FDA批准药品多维分析
FDA发布仿制药申请增补的定稿指南
全球10大畅销风湿类药物
欧盟GMP更新
监管动态
全国药品监管工作座谈会强调持续深化改革 奋力谱写新时代药监工作新篇章
7月6日,全国药品监管工作座谈会在京召开。会议以习近平新时代中国特色社会主义思想为指导,深入学习贯彻党的十九大和十九届二中、三中全会精神,落实国务院要求,总结工作,分析形势,就做好下半年和今后一个时期工作进行部署。
今年以来,全国药监系统认真贯彻落实党中央、国务院决策部署,坚持以人民为中心的发展思想,坚持稳中求进工作总基调,牢固树立“四个意识”,落实“四个最严”要求,持续深化药品医疗器械审评审批制度改革,严防严管严控安全风险,全国药品安全形势稳中向好。一是积极推进机构改革,切实做到改革和监管工作两不误、两促进。二是不断深化审评审批制度改革,境外新药上市明显加快,审评审批效率显著提升,上市许可持有人制度试点深入开展,仿制药质量和疗效一致性评价扎实推进。三是扎实开展风险防控,强化风险排查,加大现场检查力度,严惩违法犯罪行为。四是监管能力稳步提升,加快发展智慧监管、阳光监管,推进法规标准制修订工作,积极探索职业化检查员队伍建设,当选国际人用药品注册技术协调会(ICH)管理委员会成员,提升国际监管话语权。五是多措并举推进社会共治,信息公开力度不断加大,新闻宣传营造良好舆论氛围,问计于社会获得广泛支持。
为做好下半年和今后一个时期工作,李利要求:一要深入学习贯彻习近平总书记关于药监工作的系列重要指示精神,把习近平新时代中国特色社会主义思想,特别是关于药品监管工作的系列重要指示作为药监工作的理论指引和行动指南,常学常悟、深学笃行、全面落实,引领药监事业改革发展。二要扎实做好药监系统机构改革工作,严格落实党中央、国务院部署要求,在认识上要到位,在推进上要稳妥,在落实上要严格,在力量上要加强,在机制上要创新,确保顺利完成机构改革任务。三要奋力谱写新时代药监工作新篇章,坚持稳中求进工作总基调,着力创新监管方式,提升监管能力,防控安全风险,确保人民群众用药安全有效,促进药品高质量发展。强化安全意识,克服麻痹思想,加强安全隐患排查整治,提升应急处置和舆情应对能力,把问题消除在萌芽状态,把风险消灭在过程之中,确保全国药品安全形势稳定;强化服务意识,持续深化审评审批制度改革,加快进口药上市步伐,助推药品高质量发展,优化提升便民服务,每年都有志做几件增加群众和企业获得感的实事好事;强化创新意识,改革药品监管方式方法,加强职业化检查员队伍建设,大力发展监管科学,提高智慧监管水平,积极参与国际药品安全治理,不断提高审评审批、监督检验、执法检查的能力水平;强化责任意识,落实企业主体责任、地方党委政府属地管理责任、药监部门监管责任,构建群防共治的药品安全治理体系。四要加强党对药监工作的领导,全面加强党的建设,打造过硬干部队伍,持之以恒改进作风,纵深推进反腐倡廉,为药品监管事业改革发展提供坚实保证。
焦红对下半年工作做了具体部署。一要简政放权,深化审评审批制度改革。进一步健全监管法规和制度体系,加快药品医疗器械审评审批,不断完善仿制药质量和疗效一致性评价的政策措施,提高审评审批质量和效率。启动药品注射剂再评价工作,继续做好化妆品注册备案工作。二要加强监管,严厉打击违法违规行为。强化事中事后监管,聚焦疫苗、血液制品、注射剂、植入类医疗器械等高风险产品,做好进口药品医疗器械境外研发生产现场检查,推动日常检查“双随机”全覆盖,检查结果全公开;强化突出问题整治,严厉打击互联网制售假药犯罪与违法售药行为,开展中药饮片集中整治,继续开展打击无证经营与经营使用无证医疗器械专项整治行动。开展化妆品生产企业飞行检查和网络销售化妆品专项检查;强化责任落实,督促企业履行产品全生命周期责任,建立企业直接报告药品不良反应、医疗器械不良事件制度;强化监管能力提升,加强审评、检查和检验工作的质量管理,加快审评员和职业化检查员队伍建设,加强执业药师使用和管理。三要强化服务,全面推进智慧监管和阳光监管。加快推进“互联网+药品监管”,全面实行审评审批电子化,加快建立药品电子通用技术文档资料管理系统,推进医疗器械注册申请电子提交信息化系统建设。建设“药监云”,强化审评审批、检验检测、稽查执法等业务环节协同联动。继续推进阳光监管,及时公布检查、抽检、处罚等监管信息,主动回应社会关切。
来源:国家药品监督管理局
国家药品监督管理局发布《接受药品境外临床试验数据的技术指导原则》
为贯彻落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,加强对接受药品境外临床试验数据工作的指导和规范,国家药品监督管理局组织制定了《接受药品境外临床试验数据的技术指导原则》(以下简称《指导原则》)。7月10日,该指导原则正式对外发布。《指导原则》对接受境外临床试验数据的适用范围、基本原则、完整性要求、数据提交的技术要求以及接受程度均给予明确。
确保数据真实、完整、准确、可溯源
《指导原则》所涉及的境外临床试验数据,包括但不限于申请人通过药品的境内外同步研发在境外获得的创新药临床试验数据。在境外开展仿制药研发,具备完整可评价的生物等效性数据的,也可用于在中国的药品注册申报。
《指导原则》要求,申请人应确保境外临床试验数据真实、完整、准确和可溯源,这是基本原则。其数据的产生过程,应符合国际人用药品注册技术协调会(ICH)药物临床试验质量管理规范(GCP)的相关要求。申请人应确保境外临床试验设计科学,临床试验质量管理体系符合要求,数据统计分析准确、完整。
鉴于临床试验数据的完整性是接受注册申请的基本要求,《指导原则》明确在中国申请注册的产品,应提供境外所有临床试验数据,不得选择性提供临床试验数据。
数据提交有规矩
《指导原则》对于不同种类数据提交的要求进行了说明。在提交药品注册申请时,应按照《药品注册管理办法》的申报资料要求整理汇总境内外各类临床试验,形成完整的临床试验数据包。提交的数据应该包括生物药剂学、临床药理学、有效性和安全性资料数据。鼓励采用通用技术文件格式(CTD)提交。
数据质量决定接受程度
《指导原则》依据临床试验数据的质量,将接受临床试验数据分为完全接受、部分接受与不接受三种情况。
完全接受的条件包括境外临床试验数据真实可靠,符合 ICH GCP和药品注册检查要求;境外临床研究数据支持目标适应症的有效性和安全性评价;不存在影响有效性和安全性的种族敏感性因素。
若数据存在影响有效性和/或安全性的种族敏感性因素,数据外推至中国人群的有效性和安全性评价存在较大的不确定性,则为部分接受。若数据存在重大问题,不能充分支持目标适应症的有效性和安全性评价的,则属于不接受的范围。另外,对于用于危重疾病、罕见病、儿科且缺乏有效治疗手段的药品注册申请,属于“部分接受”情形的,可有条件接受。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/325800.html
来源:国家药品监督管理局
国家药品监督管理局修订清开灵注射剂和注射用益气复脉(冻干)等3个说明书
清开灵注射剂和注射用益气复脉(冻干)
根据药品不良反应评估结果,为进一步保障公众用药安全,国家药品监督管理局决定对清开灵注射剂〔清开灵注射液、注射用清开灵(冻干)〕和注射用益气复脉(冻干)说明书增加警示语,并对【不良反应】、【禁忌】和【注意事项】项进行修订。现将有关事项公告如下:
一、所有清开灵注射剂和注射用益气复脉(冻干)生产企业均应依据《药品注册管理办法》等有关规定,分别按照清开灵注射剂说明书修订要求(见附件1)和注射用益气复脉(冻干)说明书修订要求(见附件2),提出修订说明书的补充申请,于2018年8月31日前报省级食品药品监管部门备案。
修订内容涉及药品标签的,应当一并进行修订;说明书及标签其他内容应当与原批准内容一致。在补充申请备案后6个月内对已出厂的药品说明书及标签予以更换。
各相关生产企业应当对新增不良反应发生机制开展深入研究,采取有效措施做好上述品种使用和安全性问题的宣传培训,指导医师合理用药。
二、临床医师应当仔细阅读上述品种说明书的修订内容,在选择用药时,应当根据新修订说明书进行充分的效益/风险分析。
三、患者应严格遵医嘱用药,用药前应当仔细阅读上述品种说明书。
呋喃唑酮片
根据药品不良反应评估结果,为进一步保障公众用药安全,国家药品监督管理局决定对呋喃唑酮片说明书【适应症】、【禁忌】、【儿童用药】、【注意事项】等项进行修订,并增加【警示语】。现将有关事项公告如下:
一、所有呋喃唑酮片生产企业均应依据《药品注册管理办法》等有关规定,按照呋喃唑酮片说明书修订要求(见附件),提出修订说明书的补充申请,于2018年9月15日前报省级食品药品监管部门备案。
修订内容涉及药品标签的,应当一并进行修订;说明书及标签其他内容应当与原批准内容一致。在补充申请备案后6个月内对所有已出厂的药品说明书及标签予以更换。
各呋喃唑酮片生产企业应当对新增不良反应发生机制开展深入研究,采取有效措施做好使用和安全性问题的宣传培训,指导医师合理用药。
二、临床医师应当仔细阅读呋喃唑酮片说明书的修订内容,在选择用药时,应当根据新修订说明书进行充分的效益/风险分析。
三、患者应严格遵医嘱用药,用药前应当仔细阅读说明书。
重组人白介素-11注射剂
根据药品不良反应监测和安全性评估结果,为进一步保障公众用药安全,国家药品监督管理局决定对重组人白介素-11注射剂〔包括注射用重组人白介素-11、注射用重组人白细胞介素-11、注射用重组人白介素-11(Ⅰ)〕说明书【不良反应】和【禁忌】项进行修订。现将有关事项公告如下:
一、所有重组人白介素-11注射剂生产企业均应依据《药品注册管理办法》等有关规定,按照重组人白介素-11注射剂说明书修订要求(见附件),提出修订说明书的补充申请,于2018年9月15日前报省级食品药品监管部门备案。
修订内容涉及药品标签的,应当一并进行修订;说明书及标签其他内容应当与原批准内容一致。在补充申请备案后6个月内对已出厂的药品说明书及标签予以更换。
各重组人白介素-11注射剂生产企业应当对该药品相关不良反应发生机制开展深入研究,采取有效措施做好该药品使用和安全性问题的宣传培训,指导医师合理用药。
二、临床医师应当仔细阅读重组人白介素-11注射剂说明书的修订内容,在选择用药时,应当根据新修订说明书进行充分的效益/风险分析。
三、患者应严格遵医嘱用药,用药前应当仔细阅读重组人白介素-11注射剂说明书。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/229649.html、http://cnda.cfda.gov.cn/WS04/CL2050/329573.html、http://cnda.cfda.gov.cn/WS04/CL2050/329574.html
来源:国家药品监督管理局
国家药品监督管理局发布31批次药品不符合规定的通告
经江苏省食品药品监督检验研究院等6家药品检验机构检验,标示为葵花药业集团(吉林)临江有限公司等25家企业生产的31批次药品不符合规定。现将相关情况通告如下:
一、经安徽省食品药品检验研究院检验,标示为北京金崇光药业有限公司、江苏华洪药业科技有限公司、安徽华鼎堂中药饮片科技有限公司、安徽同泰佗祖堂药业有限公司、安徽亳药千草国药股份有限公司、安徽九州金诺药业股份有限公司、安徽尚德中药饮片有限公司、安徽药知源中药饮片有限公司、亳州市佰世信中药饮片有限公司、亳州市贡药饮片厂、亳州市宏大中药饮片科技有限公司、江西齐仁堂中药饮片有限公司、樟树市庆仁中药饮片有限公司、湖北金贵中药饮片有限公司、广东一信药业有限公司中药饮片厂、昆明道地中药饮片厂等18家企业生产的18批次槟榔不符合规定,不符合规定项目包括水分、黄曲霉毒素、性状、含量测定。
经山西省食品药品检验所检验,标示为江西古方原中药饮片有限公司、湖南省弘华中药饮片有限公司、安徽省金芙蓉中药饮片有限公司、广东一信药业有限公司中药饮片厂生产的4批次薄荷不符合规定,不符合规定项目包括性状、显微特征、薄层色谱。
经江苏省食品药品监督检验研究院检验,标示为葵花药业集团(吉林)临江有限公司生产的4批次炎立消胶囊不符合规定,不符合规定项目为含量测定。
经甘肃省药品检验研究院检验,标示为福州海王福药制药有限公司生产的1批次叶酸片不符合规定,不符合规定项目为有关物质。
经青岛市食品药品检验研究院检验,标示为吉林省通化博祥药业股份有限公司生产的1批次乙肝扶正胶囊不符合规定,不符合规定项目为水分。
经辽宁省药品检验检测院检验,标示为吉林恒星科技制药有限公司生产的3批次硬脂酸红霉素胶囊不符合规定,不符合规定项目为水分、溶出度。
二、对上述不符合规定药品,相关药品监督管理部门已采取查封、扣押等控制措施,要求企业暂停销售使用、召回产品,并进行整改。
三、国家药品监督管理局要求相关省级药品监督管理部门对上述企业和单位依据《中华人民共和国药品管理法》第七十三、七十四、七十五条等规定对生产销售假劣药品的违法行为进行立案调查,自收到检验报告书之日起3个月内完成对相关企业或单位的调查处理并公开处理结果。
在立案调查工作中,企业对产品真实性有异议的,可以向所在地省级药品监督管理部门提出。标示生产企业所在地省级药品监督管理部门应对该企业的生产销售情况进行调查核实,被抽样单位所在地省级药品监督管理部门应追溯问题产品来源;两地药品监督管理部门要相互配合、及时通报、一查到底,并将相关工作情况按要求及时上报。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/229691.html
来源:国家药品监督管理局
国家药品监督管理局发布角膜塑形用硬性透气接触镜及软性接触镜2项临床试验指导原则
为加强医疗器械临床试验监督管理,进一步提高注册审查质量,国家药品监督管理局组织制定了《角膜塑形用硬性透气接触镜临床试验指导原则》《软性接触镜临床试验指导原则》(见附件),现予发布。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/229671.html
来源:国家药品监督管理局
综合分析
2018年6月CDE药品审评情况分析报告
看点:
▪本月药审中心受理总量为651个(不计复审)。
▪本月国家药品监督管理局药审中心共计承办2项化药一类新药上市申请。
▪本月新增59个按一致性评价要求进行申报的受理号,包括17个注射剂受理号。
根据药智数据库最新统计,2018年6月份CDE共承办新的药品注册申请以受理号计有651个(复审除外)。
原文:https://news.yaozh.com/archive/23158.html
来源:药智网
年中盘点,2018年国内批准了这些药品
据药智数据,自2018年1月1日-6月30日,CDE共计批准了19个(受理号计,下同)新药,包括备受关注的恒瑞的硫培非格司亭注射液、正大天晴的盐酸安罗替尼胶囊、歌礼的丹诺瑞韦钠片、前沿生物的注射用艾博卫泰、万乐的注射用地西他滨等重磅药品;且还有19个进口药品,118个仿制药获得批准。
原文:https://news.yaozh.com/archive/23133.html
来源:药智网
行业信息
从审批到监管 中国医疗器械法规体系有什么变化
1989年,中国开始引入医疗器械市场准入的概念,医疗器械新产品需行政审查才可上市;1992年,借鉴欧洲的监管模式,中国启动医疗器械产品安全认证工作;1996年,原国家医药管理局发布《医疗器械产品注册管理办法》,正式规定未经注册的医疗器械不得进入市场;2000年,国务院颁布《医疗器械监督管理条例》(国务院令276号)作为中国的专项行政法规,明确规定实施医疗器械注册管理制度,中国医疗器械监管进入新的发展阶段。
中国的医疗器械监管模式借鉴了美国和欧盟的监管经验,与大多数国家一样对医疗器械实行分类管理,同时既有上市前审批,又有上市后监管。2014版《医疗器械监督管理条例》(国务院第650号令)是我国现行的医疗器械监管最高层法规文件。根据条例规定,国家对医疗器械按照风险程度实行分类管理。第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。药品监管部门负责医疗器械从研制、生产、经营到使用的全过程监管。第一类医疗器械实行产品备案管理,第二、三类医疗器械实行产品注册管理,在医疗器械上市前,所有医疗器械必须经过各级药监部门审批注册,其中第二、三类器械在首次注册时要递交临床试验报告(临床豁免目录中所列产品除外),且在产品注册时要建立质量体系并通过考核或认定。
国内外医疗器械监管模式与监管部门对比
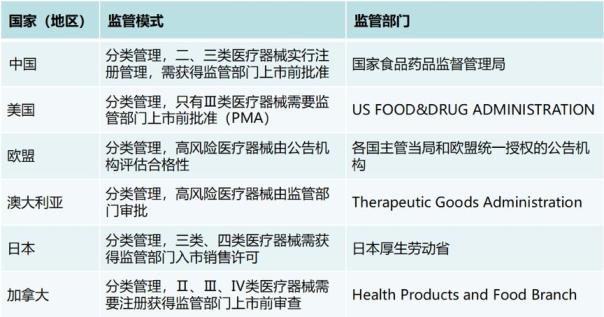
中国虽然在较短的时间里建立了较为完善的法规体系,随着我国经济社会的发展和医疗器械产业的不断壮大,在强化企业责任和创新监管手段等方面已不能完全适应形势发展的需要。从全国来看,部分医疗器械审评审批超出法定时限,审评审批涉及部门和环节过多。美国和欧盟的三类医疗器械只有10%左右,而我国的三类医疗器械比例高达20%左右,导致国家局注册审查负担沉重,存在上市前准入管理交叉重复现象,影响管理效率,为此启动了全面修订工作。
2014版《医疗器械监督管理条例》(国务院令650号)于2014年6月1日起施行,体现了国务院关于建立最严格的覆盖全过程的监管制度、深化行政审批制度改革和推进政府职能转变的精神。与旧版条例相比,新条例在完善分类管理、适当减少事前许可、加大企业责任、强化日常监督等方面做出了较大修改,主要特点如下:
1. 明确医疗器械分类管理按照风险高低程度分类;
2. 放开第一类医疗器械的经营,第二类医疗器械经营改为备案;
3. 第一类医疗器械改为备案管理,第二、三类继续实行注册审批管理;
4. “先申请生产许可证后申请产品注册证”改为“先申请产品注册证后申请生产许可证”,减少审批前行政许可事项同时降低企业成本,旧条例规定的16项行政许可降至9项;
5. 强化日常监管职责,增设医疗器械不良事件监测制度、医疗器械延续注册制度、医疗器械召回制度,通过提高处罚幅度、增加处罚种类加大违法行为的处罚力度;
6. 首次明确药监部门和卫生主管部门分别依据各自职责对医疗器械进行监督管理;
7. 明确了申请注册时应提交的资料和文件以及产品注册申请部门,规定第一类医疗器械提交的资料不包括临床试验报告。
为落实国务院2017年10月1日印发的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,国家食品药品监督管理总局于2018年再次启动了《医疗器械监督管理条例》的修订工作,并与2018年6月25日发布了条例的草案送审稿,与2014年版本相比,草案最大的变化在于除了全面落实医疗器械上市许可持有人制度外,还在于改革临床试验管理制度,第二类医疗器械原则上不需进行临床试验,此规定将使医疗器械平均提前一年左右获得批准上市。此外,监管部门还将第二类医疗器械的审评审批权限收归国家局再由国家局授权审评机构开展,推动产品的审评审批标准统一,实现审评资源合理配置。
除了从上位法加强对医疗器械的监管,我国还发布了创新医疗器械特别审批程序、医疗器械优先审批程序和免于进行临床试验医疗器械目录等措施提高医疗器械审批效率。也加强了医疗器械上市后和经营、使用环节的监管,2018年监管部门将继续加强医疗器械上市后监管工作,制定一系列规章规范性文件。包括严查网络经营和销售活动,加强医疗器械现场检查并加大处罚力度和改进抽检工作等。围绕保障医疗器械安全有效这一核心目标,监管部门科学提质提速审评审批制度,更细更严监管医疗器械全生命周期。
2018年监管部门计划制定的规章规范性文件

来源:新浪医药新闻
未来十年医疗器械行业发展前景
2030年的医疗器械行业——成为解决方案的一部分
重塑业务和运营模式,重新定位,重构价值链
“仅仅靠制造设备,然后通过分销商销售给医疗服务机构”的日子已不复存在。价值是成功的新代名词,预防是最佳的诊治结果,智能是新的竞争优势。本文探讨在2030年,医疗器械公司如何通过“三管齐下”的策略取得成功。
重塑业务和运营模式
医疗器械企业应认真审视现有的组织,通过以下方式重塑传统业务和运营模式,以适应未来的发展:
–把智能结合到产品组合和服务中,积极地影响治疗过程,并与客户、患者和消费者建立联系。
–提供超越设备的服务、超越服务的智能——真正地实现从成本到智能价值的转移。
–投资使能技术——做出正确的抉择,支持根据客户、患者和消费者(潜在患者)分别制定的多种并行业务模式——并最终为实现组织的财务目标服务。
重新定位
考虑以“由外向内”的角度为未来做准备同样重要。到2030年,外部环境将充满变数,医疗器械公司需要在新的竞争格局中重新定位,应对来自以下方面的干扰力量:
–新进入者,包括来自不相关行业的竞争者
–新技术,因为技术创新将继续比临床创新快
–新市场,因为发展中国家继续维持高速增长趋势
重构价值链
传统医疗器械的价值链将迅速演变,到2030年,企业将扮演非常不一样的角色。医疗器械企业在经历重塑业务和经营模式以及重新定位后,需要重新构建价值链,并确立它们在价值链中的位置。多种价值链“构建”方式要求企业做出根本性的战略抉择。现在已明显看到,制造商将继续与患者和消费者建立直接联系,或通过纵向一体化与医疗服务机构、甚至付款方结合起来。重建价值链的抉择并非直观的,很可能根据公司的细分市场(如器械领域、业务部和地理区域)而有所不同。由于其他企业试图重新构建价值链并实现战略目标,价值链本身将进行动态的演变,使情况变得更加复杂。然而,正确的抉择将为终端用户创造巨大的价值,并能帮助企业避免商品化的未来。
提防陷入进退两难的境地
难以承受的压力颠覆现状
医疗器械行业有望保持稳定增长,全球年度销售额预测以每年超过5%的速度增长,到2030年销售额将达到近8000亿美元1。这些预测反映了人们随着现代生活习惯病日益普遍,对创新型新设备(如可穿戴设备)和服务(如健康数据)的需求持续增长,以及新兴市场(尤其是中国和印度市场)的经济发展释放了的巨大潜能。
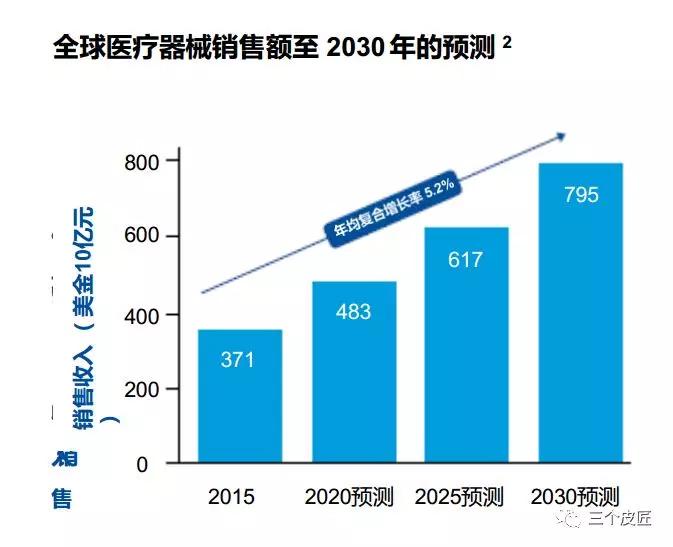
尽管前景很诱人,但无情的价格下行压力仍然如阴霾般笼罩着这个行业。全球各地政府都在力求降低医疗成本——尤其是在医疗体系中成本最高的部分:医院。他们希望在医疗器械上减少支出,同时想看到在取得更好的治疗效果方面提供更大的价值。
演变的价值链
在医疗器械未来的价值链中进行实力较量
医疗器械企业历来主要通过制造和销售产品来提供价值。然而,随着医疗体系面临的压力增大,医疗服务模式发生了根本性变化,因此,产业价值链将迎来重大变革。
在新常态下,企业需要摆脱传统制造商的角色,将服务和智能数据与产品相结合,提供整体解决方案。这就需要在价值链开展一场“实力较量”——在引进企业对消费者(B2C)模式的同时,巩固现有企业对企业(B2B)模式,并创造新模式。这场实力较量可能将包括一连串的交易活动——兼并与收购(并购)、战略联盟和合作。
重塑业务和运营模式
远不止制造设备
2030年的行业领导者将是那些与客户、患者和消费者(最终用户)建立联系、积极提供价值的医疗器械企业。企业需要结合有助降低医疗费用和改善效果的“智能”服务和解决方案,从治疗和治愈向预防转移。技术将会产生重大影响,能帮助实现预防,如果仍然有需要,还可以提供高效的微创疗法选择,减少患者留在医院的时间。
与客户、患者和消费者建立联系
为了走近终端用户,现在制造商比以往更应利用数据以及在产品中增加智能——智能很快成为了新设备价值主张的重要组成部分。数据和分析工具使企业能直接、持续地与用户建立联系,把预防的重要性放在治疗和治愈之上,让患者更好地控制自身的治疗。为了迅速提高技术能力,并有效把智能服务引入他们的产品组合中,医疗器械企业可以考虑与其他企业合作。
重新定位
新竞争格局
由于新的、非传统型市场进入者,颠覆性的技术,还有在高速发展市场崛起的、着眼于全球布局的参与者,2030年医疗器械行业的竞争格局将与当今大不相同。
新进入者
医疗保健行业正在受到颠覆性力量的影响,而这个趋势将延续到2030年。由于医疗器械行业需要以更低的成本支持更高质量的服务,未来十年将继续有来自各个行业的企业进入医疗设备行业。凭借技术的协助,这些新竞争者很可能使得现今的价值链产生冗余——正如其他平台企业所造成的改变那样。
新技术
随着令人兴奋的技术发展以前所未有的速度出现,技术既可能推动也可能颠覆医疗器械行业。正确的选择并不是直观的,对于持续不断的创新,企业需要认真评估,并进行尝试。我们认为下面五项技术将帮助把智能结合到产品组合中,在2030年将为制胜企业所用,包括:可穿戴技术、智能设备应用程序、物联网、云端数据和分析、区块链技术。我们把它们统称为“患者和消费者数据共享技术”。

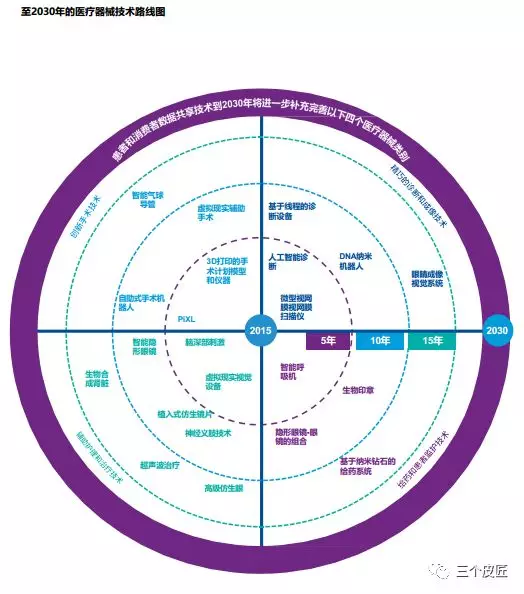
技术拓展了医疗器械企业在治疗过程的作用
新技术不仅能为医疗服务机构和患者创造效率、节省费用和带来更好的成效;还帮助医疗器械企业通过改善预防、诊断、治疗和护理,在治疗过程中发挥更广泛的作用。
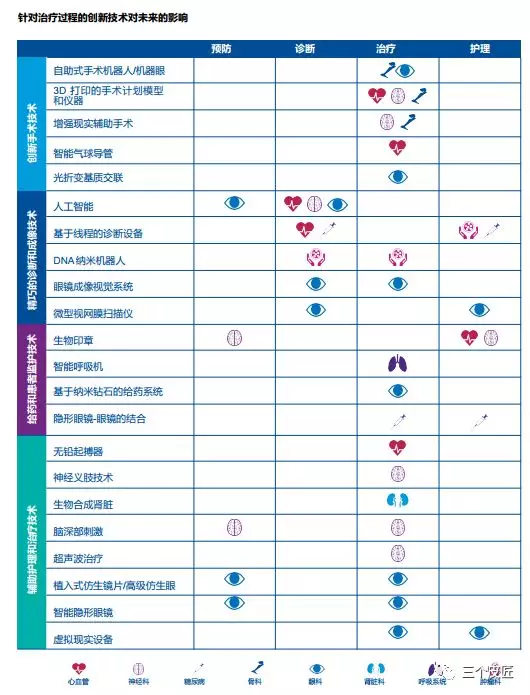
新市场
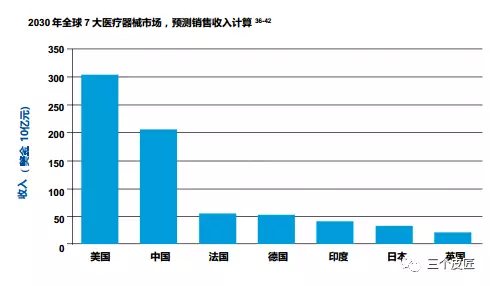
美国在2030年将继续主导医疗器械行业,销售将超过美金3,000亿元,但前五大市场还将包括中国(排名第二位,占全球市场份额超过25%,收入将超过美金2,000亿元32)和印度(排名第五,收入将超过400亿美元33)。受医疗改革、地方政府的激励措施和对医疗的总体需求增长的推动3435,中国和印度的增长速度已经是整体市场的两倍。这两个国家也在快速地发展成为创新中心——印度已经被誉为廉价工程技术的全球中心,制造了许多具有全球市场潜力的本土(和低成本)器械。
重构价值链
在未来的价值链中占一席位
随着市场的快速发展,现有的市场参与者需要考虑在2030年的医疗器械价值链中如何定位,以避免成为单纯的商品供应商。就连最强、最大的企业也会容易受到颠覆市场的新进入者、环球竞争和技术跃进的威胁。医疗器械制造商要始终专注于他们为未来医疗体系带来的价值,同时需要考虑开展大胆而必要的实力较量,重新构建价值链。
企业的不同分部可能需要不同的价值链模式,因此需要对各种选择进行仔细评估以获得全面认识。我们认为,一直到2030年会存在几种模式,包括:
1.建立新的B2C模式
2.加强和巩固B2B模式
3.在价值链中做大
在2030年保持领先地位
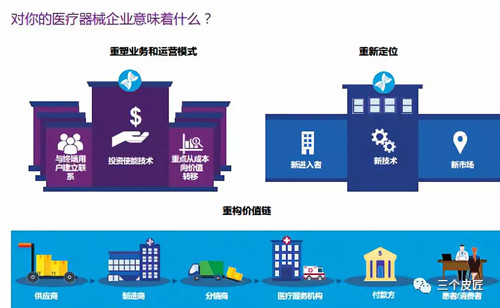
要开启迈向2030年的旅程,实现“重塑业务和运营模式–重新定位–重构价值链“的战略,你能做些什么?为了在未来构建成功的价值链模式,医疗器械企业应考虑以下建议:
确定价值主张
明智投资
协作和建立生态系统
采用灵活、模块化的组织架构
别让过去羁绊未来的发展
来源:药智网
国外信息
无菌企业自检清单——FDA检查员手册的168个问题
美国FDA的行业指南被业界广泛学习,但其实FDA还有很多检查和合规相关的指南,识林已经进行完整收集,并翻译了部分关键指南。
来源:识林
美国FDA批准药品多维分析
分析5月份FDA批准的药品,从治疗领域、申请类型、提交分类等方面介绍产品。
在所有的104个产品批文中,按照系统划分,涵盖了12个系统。生殖泌尿系统及性激素(16条)、消化系统及代谢(15条)、神经系统(13条)三类药物批文数最多。
原文:https://news.yaozh.com/archive/23138.html
来源:药智网
FDA发布仿制药申请增补的定稿指南
7月4日,美国 FDA 发布题为《ANDA 申请递交 — GDUFA 下的 ANDA 增补》定稿指南。与去年 10 月发布的指南草案一致,定稿指南舍弃了 GDUFA I 复杂的分层级增补分类系统,而是根据 GDUFA II 目标函提供了一种不那么繁琐且更直接的增补分类。指南提供了关于这一变化的背景,定义了重大、微小和 FDA 未要求申请人自愿提交的增补,讨论了新目标日期的适用情况,并提供了对某些分类复议的流程。另外定稿指南还包括一个附录列出了仿制药办公室(OGD)和药品质量办公室(OPQ)认为的重大缺陷,以帮助申请人更好地理解其增补对应的目标日期和分类。
指南指出,“本指南中描述的增补类型和审评目标仅适用于已接收并将进行评估的递交(即,审评目标不适用于等待立卷审查的递交)。信息请求(IR)和学科审评函(DRL)既不会停止审评时间进展,也不会添加到 GDUFA II 目标中”,除非在申请人的回复中对这些请求的回复包含额外的新信息。
目标日期对于 ANDA 申请人来说是最重要的,指南对于 ANDA 和需事先批准的补充申请(PAS)的增补提供了与指南草案中基本相同的时间框架,如下表所示:
表 1:ANDA 重大和微小增补的绩效目标总结

表 2:PAS 重大和微小增补的绩效目标总结

仿制药生产商请一定要认真研究这些表格 , 以确保对自己所提交申请的目标日期心中有数。如果你的增补递交不在表中所列的 90% 范围内,你的递交出了什么问题仍有待观察。请仔细审阅指南 , 因为其它情况(例如,GDUFA II 之前的增补、暂时批准申请的增补以及延期增补等)对于你了解自己的增补状态都至关重要。
来源:识林
全球10大畅销风湿类药物
目前,风湿病是全球制药企业涉足较大的疾病领域之一,因此针对其治疗的药物销量并不逊色居于畅销榜第一的肿瘤药。据统计,2017年全球10种最畅销的风湿病药物(详见下图)的总收入接近570亿美元。
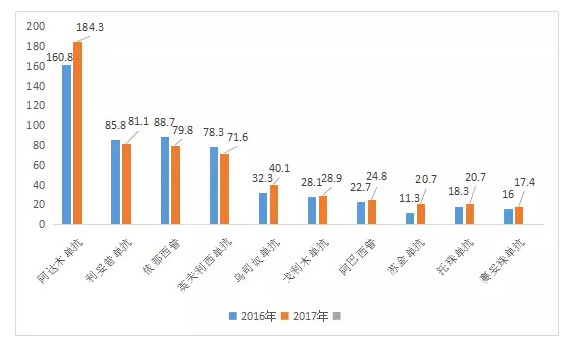
2016-2017年全球十佳风湿药的销售额情况(单位:亿美元)
TOP1 阿达木单抗(修美乐)
荣登2017年风湿药物畅销榜第一的属艾伯维旗下的典型产品阿达木单抗,该药是一种肿瘤坏死因子(TNF)阻断剂,可用于治疗类风湿样关节炎(RA)、幼年特发性关节炎(JIA)、银屑病关节炎(PsA)、强直性脊柱炎(AS)、克罗恩氏病(CD)等相关适应症。阿达木单抗的美国专利于2016年到期,欧洲专利也将于今年到期。
阿达木单抗在2017年的销售金额高达184.3亿美元,同比增长14.60%。
阿达木单抗在2009年6月获国家食品药品监督管理局批准临床,且于2010年8月,正式进入中国市场,一度处于“垄断”地位。但随着中国生物药企业的研发实力不断更迭,目前已有通化东宝生物科技有限公司在2017年2月获阿达木单抗注射液的临床批件,华兰基因工程有限公司、正大天晴药业集团股份有限公司于2017年3月获得阿达木单抗注射液的新药临床批件。若三家企业生产的阿达木单抗注射液可以成功在国内上市,将有望打破阿达木单抗依赖进口的僵局。
TOP2 利妥昔单抗(美罗华)
利妥昔单抗为罗氏的重磅药之一,是一种指向CD20溶细胞抗体适用于治疗非霍奇金淋巴瘤(NHL)、慢性淋巴细胞白血病(CLL)、类风湿样关节炎(RA)与甲氨蝶呤联用对一种或更多TNF拮抗剂治疗反应不佳的有中度至严重获得性RA成年患者、Wegener氏肉芽肿(WG)和显微镜性多发性血管炎(MPA)等症。
利妥昔单抗在2017年销量达81.1亿美元,同比减少5.47%,略有下滑的趋势。
利妥昔单抗在2002年12月进入中国,之后的十年,上海罗氏制药有限公司一家独大,占据CLL、RA的大部分用药市场。近两年,生物创新药市场需求的剧增激发了大批企业的研发热情。国内已有华兰基因工程有限公司、正大天晴药业集团股份有限公司、上海复宏汉霖生物制药有限公司三家企业获得利妥昔单抗注射液的临床批件,进度较快的为上海复宏汉霖,去年12月该公司的利妥昔单抗被纳入优先审评程序,且于同月申报上市。此外,为了减轻患者负担,利妥昔单抗还在2017年被纳入国家医保药品目录。
TOP3 依那西普
依那西普由惠氏(辉瑞子公司)和安进联合开发,于1998年11月2日获得美国FDA批准,该药是一种肿瘤坏死因子(TNF)阻断剂,用于治疗类风湿样关节炎(RA)、年龄2岁或以上多关节幼年特发性关节炎(JIA)患者、银屑病关节炎(PsA)、强直性脊柱炎(AS)、斑块性银屑病(PsO)等疾病。
依那西普在2017年的销售额达79.8亿美元,低于2016年10个百分点。
依那西普在2010年2月26日进入中国市场,由勃林格殷格翰在中国市场销售,商品名为Enbrel®。2017年12月13日,辉瑞的依那西普注射液获得中国国家食品药品监督管理总局(CFDA)批准。此外,据药智数据,目前国内药企均无进行依那西普的相关申报记录。但值得提及的是,西藏自治区、宁夏回族自治区两地已将此款进口药纳入医保乙类药品(2017年版)。
TOP4 英夫利西单抗
英夫利西单抗由杨森(强生的子公司)研发,主要用于治疗克罗恩病、溃疡性结肠炎、类风湿样关节炎、强直性脊柱炎、银屑病关节炎、斑块性银屑病等疾病。它于1998年8月24日获得美国食品药品管理局(FDA)批准,由杨森、默克和田辺三菱在美国、欧洲、日本和中国共同销售,商品名为Remicade。
英夫利西单抗在2017年销量达到71.6亿美元,同比下降8.55%。
注射用英夫利西单抗于2006年进入中国市场(各地挂网价范围均在5100-6000元),强生是该药唯一的产、销企业。此外,英夫利西单抗在2017年进入国家医保药品目录,随后,甘肃省及西藏自治区将其纳入乙类医保药品(2017年版)。
TOP5 乌司奴单抗
乌司奴单抗是由美国强生研发和销售,作为银屑病的经典标准用药。该药是一种人类白介素-12和-23拮抗剂用于治疗中度至严重斑块银屑病(Ps)(光治疗或全身治疗备选者)、活动性银屑病关节炎(PsA)的成年患者(18岁或以上)。且为强生的主要产品之一,每年创收约上亿美元,且逐年剧增。
乌司奴单抗在2017年为强生带来40.1亿美元的收益,首次突破40亿美元,同比增长24.14%。
强生的乌司奴单抗注射液在2017年11月已获得进口注册证。国内药企暂无任何申报记录,强生享有“独家专利”。
TOP6 戈利木单抗
戈利木单抗最早是强生与先灵葆雅联合研发的一款药物,于2013年获美国FDA批准用于中、重度活动性类风湿性关节炎(RA)的治疗。而在此之前,杨森制药将戈利木单抗作为风湿性关节病的治疗药引进日本,并在2006年8月与田边三菱制药公司缔结共同生产、销售的合作。因此,戈利木单抗的盈利由三家企业瓜分(强生、田边三菱制药份额占比最多,默克次之)。而默克制药能分得一杯羹,主要得利于2009年以430亿美元的价格收购竞争对手先灵葆雅,同时收获了先灵葆雅与强生联合研发的戈利木单抗(Golimumab)。
戈利木单抗2017年的销售额达28.9亿美元,同比增长2.84%。
戈利木单抗于2018年1月获得国家食药监总局的批准,是中国首个获批的每月皮下注射一次的抗风湿生物制剂。
TOP7 阿巴西普
阿巴西普是由百时美施贵宝研发的生物药之一,已在美国、欧洲和日本上市。该药是一种选择性T细胞共刺激调节剂,主要用于1种或多种缓解病情抗风湿药(DMARD),如甲氨蝶呤、肿瘤坏死因子(TNF)阻断剂治疗但应答不足的中、重度活动性类风湿关节炎成年患者。
阿巴西普是百时美施贵宝的热销产品之一,2017年创收24.8亿美元,同比增长9.25%。
2013年7月,百时美施贵宝和先声药业联合宣布,双方将在中国携手研发上市用于治疗风湿性关节炎的生物制剂阿巴西普,并且先声药业将负责阿巴西普针剂在中国的审批及审批前的研发事务。一旦阿巴西普成功获批,双方共同分享其市场收益。据药智数据,江苏先声药业有限公司的阿巴西普在2015年11月已获临床批准。
TOP8 苏金单抗
苏金单抗最早是由诺华公司出品开发,并于2015年1月在美国获批上市,随后相继进入瑞士、加拿大、英国、欧盟、日本、香港、新加坡、菲律宾、印度等国的银屑病用药市场。该药是一种人白介素-IL-17A拮抗剂,适用为全身治疗或光治疗备选者的中度至严重斑块性银屑病成年对治疗。
苏金单抗在2017年为诺华公司创造20.7亿美元,同比增长83.18%,为此次风湿药畅销实力榜中增速最快的,也是诺华公司独有的产品之一。本品在国内暂无临床或上市等申报记录。
TOP9 托珠单抗
托珠单抗主要用于使用抗风湿药物(DMARDs)期间,而应答不足的中到重度活动性类风湿关节炎的成年患者。由于临床适应症的新拓展,该药在2017年8月成为首个获得FDA批准用于治疗CAR-T疗法引起的严重或致命的细胞因子突释综合征(CRS)的药物。
托珠单抗在2017年销售额达20.7亿美元,同比增长13.11%。
2013年3月,托珠单抗在中国获批第一个适应症类风湿关节炎(RA),用于治疗应答不佳的中重度活动型,主要用于成年患者。2016年11月托珠单抗得到sJIA适应症(一种自身免疫性疾病,是幼年特发性关节炎(JIA)中最严重的一种亚型,严重影响儿童的生长发育,致残率和死亡率都很高)的获批。而这两个适应症使其拥有风湿病领域的绝对优势。
TOP10 赛妥珠单抗
赛妥珠单抗由UCB公司研发,是一种肿瘤坏死因子(TNF)阻断剂,适用于克罗恩病对常规治疗反应不佳的有中度至严重活动性疾病的成年患者中,旨在减轻体征和症状、维持临床反应;治疗有中度至严重活动性类风湿样关节炎的成年患者;治疗有活动性银屑病关节炎成年患者。该药在2008年获FDA批准用于克罗恩病、类风湿性关节炎、银屑病关节炎及强直性脊柱炎的治疗。
赛妥珠单抗在2017年销量大17.40亿美元,同比增长8.75%,其最大的受益者为优时比公司(比利时的一家跨国生物制药企业)。现目前,本品在国内无临床或上市的申报记录。
来源:药智网
欧盟GMP更新
近日,欧洲药品管理局(EMA)官员Roberto Conocchia和意大利前GMP检查员介绍了有关EU GMP的修订计划和进展:

﹒Detailed guidelines on GMP for Investigational Medicinal Products (IMPs) – Published
《临床试验用药GMP指南》(IMPS):已发布
﹒Detailed Guidelines on GMP for Advanced Therapy Medicinal Products (ATMPs) - Published (Entered into force May2018).
《先进治疗药品GMP》(ATMPS):已发布,2018年5月生效
﹒Chapter 1 (Pharmaceutical Quality System) - Work starting in 2018
第1章(药品质量体系):2018年启动修订
﹒Annex 1 (Manufacture of Sterile Medicinal Products) - In progress
附录1(无菌药品的生产):修订中
The IWG is currently working on the finalisation of Annex 1. An impressive number of 6200 lines of comments addressed to the document have to be worked through. This work should be finished by December 2018. A publication is expected for March 2019.IWG目前正在完成附录1的最终工作。需要考虑对该文件的6200条评论意见。这项工作将于2018年12月完成。新指南预计2019年3月发布。
﹒Annex 2 Manufacture of Biological active substances and Medicinal Products for Human Use (into operation since 26 June 2018)
附录2 -人用生物活性物质和药品的生产(自2018年6月26日起生效)
Annex 2 is no longer applicable to Advanced Therapy Medicinal Products to which applies the Commission guideline on Good Manufacturing Practice for Advanced Therapy Medicinal Products, published in Part IV of Eudralex Volume 4 and operational as of 22 May 2018.
附录2不再适用于先进治疗药物产品,先进治疗药物产品相关要求见 Eudralex第4卷第IV部分, 2018年5月22日实施。
﹒Chapter 4 and Annex 11 - Work starting in 2018
第4章和附录11:2018年启动修订
﹒GMP for importers of medicinal products ("Annex 21) - Expected public consultation June 2018
药品进口商GMP(附件21):预计2018年6月发布征求意见稿
﹒Q&A on implementation of risk-based prevention of cross-contamination in production and 'Guideline on settinghealth-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities – Published
基于风险防止药品生产中交叉污染以及“共用设施中不同药品生产风险识别所用基于健康的暴露限设定指南”实施问答
﹒Annex 17 Parametric release (Deadline for coming into operation: 26 December 2018)
附录17 参数放行(2018年12月26日生效)
﹒Annex 13 Manufacture of Investigational Medicinal Products
附录13 临床试验用药品的生产
Detailed Commission guideline of 8 December 2017 on the good manufacturing practice for investigational medicinal products pursuant to the second paragraph of the Article 63(1) of Regulation (EU) No 536/2014 (applicable as from the date of entry into application of Regulation (EU) No 536/2014 on Clinical Trials)
2017年12月8日关于根据法规(EU)No 536/2014第63(1)条第2段开展的临床试验用药品GMP指南(自条例(EU) No 536/2014生效之日起适用)
来源:GMP办公室