本 期 要 目
[监管动态]
国家药品监督管理局发布批准注册80个医疗器械产品公告
国家药品监督管理局发布28批次化妆品不合格的通告
国家药品监督管理局发布注销黄瑞香注射液(国药准字Z14021011)药品批准证明文件的公告
国家药品监督管理局发布医疗器械规范性文件(1998—2013年)清理结果的公告
国家药品监督管理局发布36批次化妆品不合格的通告
[综合分析]
2018年Q3纳入药品优先审评品种分析
一致性评价:292个BE试验已完成,69个药品或将进入申报
政策驱动下的我国儿童药研发现状
[国外信息]
EDQM:OMCL发布3个沙坦中NDMA检测方法
FDA:原料药批准后变更(指南草案)
FDA发布药品调配外包设施相关要求的指南
FDA发布两份指南为药品申请的高质量递交提供建议
FDA药品申请实时审评成为现实
监管动态
国家药品监督管理局发布批准注册80个医疗器械产品公告
2018年8月,国家药品监督管理局共批准注册医疗器械产品80个。其中,境内第三类医疗器械产品47个,进口第三类医疗器械产品20个,进口第二类医疗器械产品13个(具体产品见附件)。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/330334.html
来源:国家药品监督管理局
国家药品监督管理局发布28批次化妆品不合格的通告
经重庆市食品药品检验检测研究院等检验,标示为广州市白云区莱丹精细化工厂等21家企业生产的28批次化妆品不合格(见附件1)。现将有关情况通告如下:
一、涉及的标示生产企业、不合格产品为:广州市白云区莱丹精细化工厂生产的索菲雅染发膏(自然黑),广州市卡迪娜化妆品厂生产的卡迪娜染发膏(金黄色),广州歌秀化妆品有限公司生产的愈美染发膏,广东瑞虎精细化工有限公司生产的瑞虎染得快染发膏(栗棕色),广东圣薇娜精细化工有限公司生产的然你魅4/66圣薇娜彩色焗油(红色),广州丽臣精细化工有限公司生产的丽臣染发膏(板栗色),广州市金栢丽保健品有限公司生产的干恋染发膏(自然黑),广州市兰姿化妆品有限公司生产的海维斯染发焗油膏(自然黑),广州市凯维斯化妆品有限公司生产的凯维斯染发膏(紫红色),广州威妮雅化妆品有限公司生产的金竹堂染发膏(酒红),鹤山市邦丽精细化工有限公司生产的梦佩丝染发膏(红色),广州温雅日用化妆品有限公司生产的温雅漾采染发焗油(5B)咖啡色,中山佳丽日用化妆品有限公司生产的利威丝染发霜(栗棕色),广州市美度化妆品有限公司生产的美度染发膏(棕黑色)44/0和美度染发膏(酒红色)S-5/64,广州市白云区圣迪雅化妆品厂生产的益容堂染发膏(葡萄红)和益容堂染发膏(自然黑),广州市汉邦化妆品有限公司生产的昌义润黑露染发膏(自然黑色)、昌义汉邦染发膏(自然黑色)和汉邦染发膏(棕色5.4),广州嘉瀛化妆品有限公司生产的嘉瀛染发膏(棕色)和嘉瀛染发膏(红色),大连河原日用化学品有限公司(北京顺捷彩悦化妆品有限公司监制)生产的彩蕴焗油染发膏天然黑,北京日光旭升精细化工技术研究所生产的珍草堂彩染焗油膏(自然黑色)和珍草堂彩染焗油膏(栗棕色),浙江章华保健美发实业有限公司生产的章华汉草焗油染发霜,广州市彩琳日用化工有限公司生产的彩琳®美白保湿防晒露SPF30+PA+++。
其中,经生产企业所在地食品药品监管部门现场核查,标示广州威妮雅化妆品有限公司生产的金竹堂染发膏(酒红),广州嘉瀛化妆品有限公司生产的嘉瀛染发膏(棕色)和嘉瀛染发膏(红色),浙江章华保健美发实业有限公司生产的章华汉草焗油染发霜等相关批次产品,标示生产企业否认为该企业生产。
二、上述不合格产品及相关企业违反了《化妆品卫生监督条例》《化妆品标识管理规定》等相关规定。国家药品监督管理局要求广东、辽宁、北京、浙江省(市)食品药品监督管理局核实后依法督促相关生产企业对已上市销售相关产品及时采取召回等措施,立案调查,依法严肃处理;要求重庆、青海、黑龙江省(市)食品药品监督管理局责令相关经营单位立即采取下架等措施控制风险,对涉嫌假冒的产品,要深查深究其进货渠道,对违法违规行为,依法予以查处,涉嫌犯罪的依法移交公安机关。上述省级食品药品监督管理局自通告发布之日起3个月内公开对相关企业或单位的处理结果,相关情况及时在国家化妆品抽检信息系统中填报并报告国家药品监督管理局。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/330358.html
来源:国家药品监督管理局
国家药品监督管理局发布注销黄瑞香注射液(国药准字Z14021011)药品批准证明文件的公告
根据《中华人民共和国药品管理法实施条例》和《药品注册管理办法》的有关规定,国家药品监督管理局决定注销黄瑞香注射液(国药准字Z14021011)药品批准证明文件。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/330393.html
来源:国家药品监督管理局
国家药品监督管理局发布医疗器械规范性文件(1998—2013年)清理结果的公告
根据中共中央、国务院印发的《法治政府建设实施纲要(2015—2020年)》的要求,为做好药品监管法律制度“立改废释”工作,全面推进依法行政,国家药品监督管理局组织对1998—2013年医疗器械规范性文件进行了清理,并决定废止和宣布失效一批规范性文件。现将《国家药品监督管理局继续有效的医疗器械规范性文件目录(1998—2013年)》(附件1)和《国家药品监督管理局废止和宣布失效的医疗器械规范性文件目录(1998—2013年)》(附件2)予以公布。
对上述予以废止或者宣布失效的规范性文件,除另有明确规定外,均不涉及过去根据这些文件所作出处理决定的效力。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/330394.html
来源:国家药品监督管理局
国家药品监督管理局发布36批次化妆品不合格的通告
经辽宁省药品检验检测院等检验,标示为广东圣薇娜精细化工有限公司等27家企业生产的36批次化妆品不合格(见附件1)。现将有关情况通告如下:
一、涉及的标示生产企业、不合格产品为:广东圣薇娜精细化工有限公司生产的圣薇娜彩色焗油(棕色),广州市佳桐化妆品有限公司生产的艾贝尔植物酵素黑发乳,广州市三荣精细化工有限公司生产的三荣染发膏(紫色),广州市发爵士精细化工有限公司生产的发爵士染膏(自然黑),广州富品生物科技有限公司生产的雅缇染发膏(黑色),广州市丹缇化妆品有限公司生产的迪丝柔丹缇染发膏(自然黑),广州市伊春化妆品有限公司生产的汉丰染发膏(黑色),广州起秀化妆品有限公司生产的韩佰焗油染发膏白发王4/0棕色,广州市新海岸精细化工有限公司生产的新海岸染发膏(自然黑),广州市维珍妮化妆品有限公司生产的维珍妮染发膏(自然黑),广州宜合三口化妆品有限公司生产的3D极爱染色膏 凤风染发膏(棕色),广东顺德黛尼美日用化妆品科技有限公司生产的黛尼美染发膏(C-5/67葡萄紫色),肇庆市凯捷科技有限公司生产的凯捷染发膏(闷青色),广州市白云区伊多娜化妆品厂生产的茶果清水黑油,广州德洛莉丝生物科技有限责任公司生产的韩金靓®Deluolisi德洛莉丝染发霜(黑色),广州市靓鑫精细化工有限公司生产的靓鑫染发膏(自然黑),广州俪凝化妆品有限公司生产的俪缇染发膏(自然黑),广州市韩妃化妆品有限公司生产的韩妃染发膏(紫红色),广州市澳伦化妆品有限公司生产的怡美姿染发膏,北京三精日化有限公司生产的精彩焗染膏(浓情醉紫),济南雪豹邦仕化妆用具有限公司生产的雪豹牌染发膏(自然黑),大连河原日用化学品有限公司(北京顺捷彩悦化妆品有限公司监制)生产的彩蕴焗油染发霜11号和彩蕴速染焗油膏自然黑,广州市景红达精细化工有限公司生产的景红达染发膏(褐色)、景红达烫发液(热敷型)(蓝炫小星蚕丝蛋白烫)、景红达烫发液(热敷型)发朵朵氨基酸抛光弹性保湿香水烫、景红达烫发液(热敷型)优能活性弹力水波纹烫、景红达烫发液(热敷型)(弹力红姿雅保湿抛光烫发水)、景红达烫发液(热敷型)(弹力绿姿雅保湿抛光烫发水)、景红达烫发液(热敷型)标美氨基酸芳香特效香水烫、景红达烫发液(热敷型)韩秀蛋白修复香水烫和景红达烫发液(热敷型)草本植物保湿精华生化烫,广州市鑫姿化妆品有限公司(委托方:广州包氏化妆品有限公司)生产的鑫姿烫(曲、直)发液,广州艾婷化妆品有限公司生产的可卡可娜一洗烫,广州市鑫锦化妆品有限公司生产的鑫锦烫发水,广州贝嘉欣化妆品有限公司生产的贝嘉欣烫发水。
其中,经生产企业所在地食品药品监管部门现场核查,标示北京三精日化有限公司生产的精彩焗染膏(浓情醉紫),标示生产企业否认为该企业生产。
二、上述不合格产品及相关企业违反了《化妆品卫生监督条例》《化妆品标识管理规定》等相关规定。国家药品监督管理局要求广东、北京、辽宁、山东省(市)食品药品监督管理局核实后依法督促相关生产企业对已上市销售相关产品及时采取召回等措施,立案调查,依法严肃处理;要求辽宁、甘肃、广西、湖南、吉林、四川省(区)食品药品监督管理局责令相关经营单位立即采取下架等措施控制风险,对涉嫌假冒的产品,要深查深究其进货渠道,对违法违规行为,依法予以查处,涉嫌犯罪的依法移交公安机关。上述省级食品药品监督管理部门自通告发布之日起3个月内公开对生产销售不合格化妆品相关企业或单位的处理结果,相关情况及时在国家化妆品抽检信息系统中填报。
原文:http://cnda.cfda.gov.cn/WS04/CL2050/330412.html
来源:国家药品监督管理局
综合分析
2018年Q3纳入药品优先审评品种分析
2018年7月至9月,我国共有83个受理号的药品被纳入优先审评。
我国现行的药品优先审评制度是在推进药品审评审批改革、解决药品审评积压的背景下确立实施的,自2016年7月国家药审中心发布《“首仿”品种试行优先审评评定的基本原则》以来,我国已经共有18类药品被列入优先审评支持范围。
1、药品优先审评进展概况
药品优先审评制度实施以来,全国已经共有660个受理号的药品被纳入了优先审评,包括415个国产药品和245个进口药品,391个新药申请和269个仿制药申请。
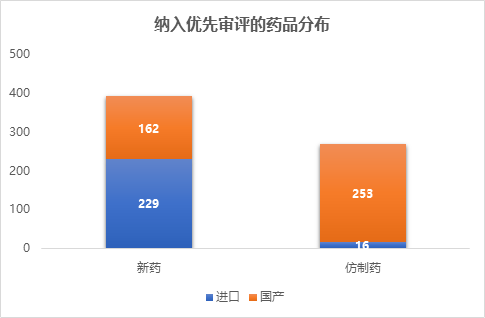
2018年第三季度,共有83个受理号的药品被纳入优先审评,较第二季度减少了17%。至此,2018年前三季度共有227个药品纳入优先审评,较去年同期增加了44.6%。从历年各季度纳入优先审评的药品数量上来看,除了开始实施的2016年情况特殊,2018年相比于上一年,纳入的药品数量有了大幅提升,仅前三季度的总数量就已经逼近了2017年全年的数量。
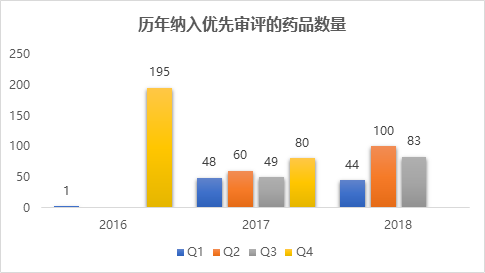
2018年Q3纳入品种分布
2018年Q3被纳入优先审评的药品中,新药申请32个,包含24个化药、7个生物制品和1个中药,仿制药51个,均为化药,因此化药总数为75个,占全部的90%;国产药品68个,进口药品申请15个,包含9个临床申请和6个生产申请。
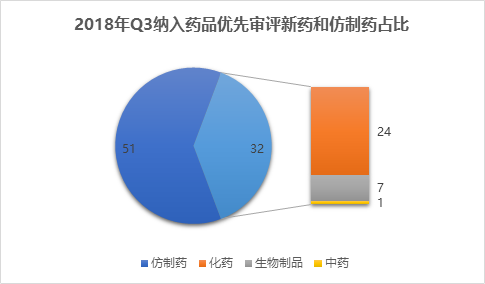
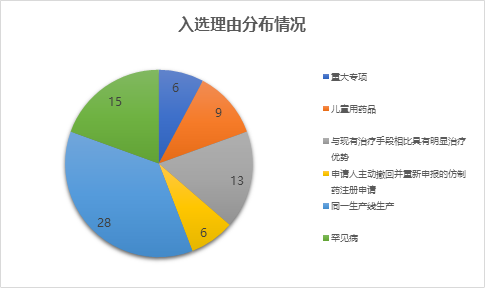
从入选理由来看,以“同一生产线生产并已有产品在美国、欧盟国家等上市”入选名单的品种最多,“罕见病”和“与现有治疗手段相比具有明显优势”位列二三。
从注册申请人的地域分布情况来看,68个国产药品最主要分布在广东、江苏、浙江和山东,北京仅有4个,上海在三季度则并无药品被纳入优先审评。
从申请人和品种的分布情况来看,齐鲁制药及其海南公司共有10个药品被纳入优先审评,总数上排在第一,包括了2个规格的注射用培美曲塞二钠、4个规格的他达拉非片和4个规格的依托考昔片;紧随其后的是爱可泰隆医药贸易(上海)有限公司8个规格的进口药Selexipag片和1个波生坦分散片;排在后面的是东阳光药(6个)和海正药业(5个),以及均有4个药品被纳入的吉林津升制药和豪森药业。
2、重点药物简介
Selexipag片:爱可泰隆的Selexipag片是以国际多中心临床试验数据申请进口的罕见病药物,用于治疗目前致病因素尚不清楚、也尚无治愈手段的肺动脉高压(PAH)。Selexipag是爱可泰隆的第三款治疗PAH的药物,在其之前的马昔腾坦(Opsumit)和波生坦(Tracleer)均给爱可泰隆带来了长期的增长,波生坦片更是这家欧洲生物技术公司的支柱产品,占据了公司80%的销售额,。有瑞士信贷银行的分析师预测Selexipag这款药物到 2020 年的销售额可以达到 5.75 亿瑞士法郎(6.55 亿美元)。
KW-136胶囊:NS5A抑制剂KW-136胶囊是凯因科技具有自主知识产权的丙肝一类新药,作为重大专项被纳入药品优先审评,其与与索磷布韦片联合使用成为国内首个全口服、全基因型的丙肝根治方案。
马来酸艾维替尼:马来酸艾维替尼是由艾森医药自主研发的国内首个第三代EGFR抗肿瘤靶向抑制剂,也是原创国产1.1类新药,拥有全球化合物专利,用于治疗具有EGFR突变或耐药突变的非小细胞肺癌,作为十三五国家“重大新药创制”科技重大专项而被纳入药品优先审评。
重组人乳头瘤病毒16/18型双价疫苗(大肠杆菌):厦门万泰与厦门大学联合研制的重组人乳头瘤病16/18型双价疫苗是国内首支进入临床试验的国产重组人乳头瘤病毒疫苗,目前国产HPV疫苗的研发竞争非常激烈,有包括厦门万泰、成都生物制品研究所、上海泽润生物、上海博唯生物等在内的多家公司的6款产品处于临床试验阶段,而厦门万泰获得优先审评的二价HPV疫苗,无疑将成为有望最快上市的国产HPV疫苗。
格隆溴铵注射液:格隆溴铵注射液是一种抗胆碱能药物。目前国内仅有格隆溴铵片获批上市,尚未有该药品注射剂获批上市。2012年至今,国内有恒瑞医药、成都苑东和广东嘉博制药等8家企业获得格隆溴铵注射液临床批件,目前仅有恒瑞医药提交上市申请。2017年格隆溴铵注射液全球销售额约为1.89亿美元。
盐酸伐地那非片:盐酸伐地那非片是一种PDE5抑制剂,用于治疗男性勃起功能障碍,其原研由拜耳和GSK联合开发。科伦药业是国内首家盐酸伐地那非片通过生物等效性研究(BE)并以与原研质量一致性标准申报生产的企业。
来源:火石创造网
一致性评价:292个BE试验已完成,69个药品或将进入申报
随着“仿制药质量和疗效一致性评价”政策的出台,临床试验的关注度不断提高。截止2018年9月19日,平台获得CTR号的仿制药一致性评价BE试验685项,其中292项显示已完成。
685个已登记的仿制药一致性评价BE试验主要格局如下:
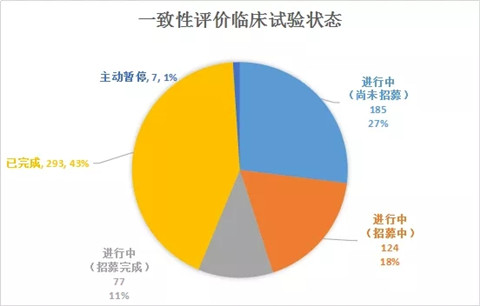
本文将重点介绍已完成的292个BE试验,同时结合注册与受理情况进行简要的分析。
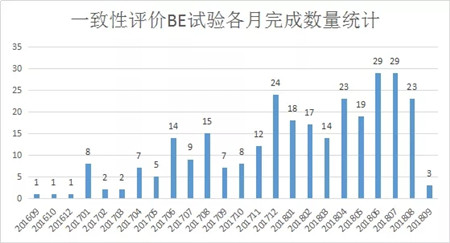
01 已完成的一致性评价BE试验统计
通过统计2016年至2018年的临床试验,对比药品上市信息和注册信息,筛选出了用于仿制药一致性评价的BE试验。截止2018年9月19日,已完成的BE试验总计达到292个。下图为各月的仿制药一致性评价BE试验完成(终止)数量统计图:
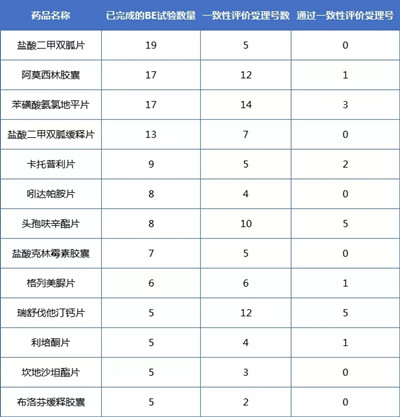
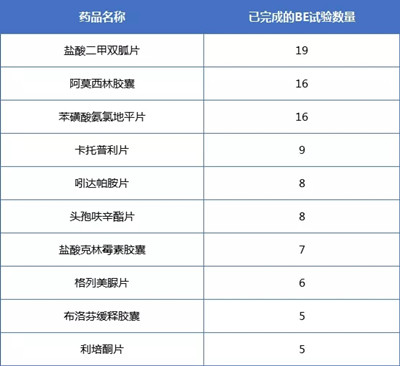
02 已完成的BE试验最受关注的药品
对一致性评价已完成的BE试验的药品统计发现,截止2018年9月19日完成BE试验最多的药品为盐酸二甲双胍片,达到19个登记号;其次是苯磺酸氨氯地平片和阿莫西林胶囊,分别为17个。结合仿制药一致性评价注册申报情况可以看到,尽管盐酸二甲双胍片BE试验很多,一致性评价的受理号却只有5个,且还没有通过的。已完成BE试验的品种排行如下:
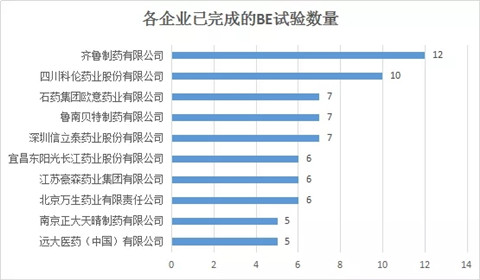
03 各企业已完成的BE试验数量
总的来说,截止2018年9月19日,共有154家企业的仿制药一致性评价BE试验已经完成,91家企业只有1个已完成的BE试验。齐鲁制药有限公司已完成的BE试验数量最多,登记号达到12个;其次是四川科伦,登记号为10个。TOP10企业几乎为国内大型药企,可见,大型药企对仿制药质量和疗效一致性评价重视度更高,且表现出较强的实力。
04 已完成的BE试验中289目录情况
数据显示,截止2018年9月19日,已完成一致性评价BE试验的登记号中,属于289目录的临床试验为176个,占整体的60.27%。
简单罗列了一下289目录一致性评价已完成BE试验的最受欢迎的药品,可以看出基本和整体完成BE的品种相同,也就是说,目前完成BE试验最多的药品基本属于289目录。
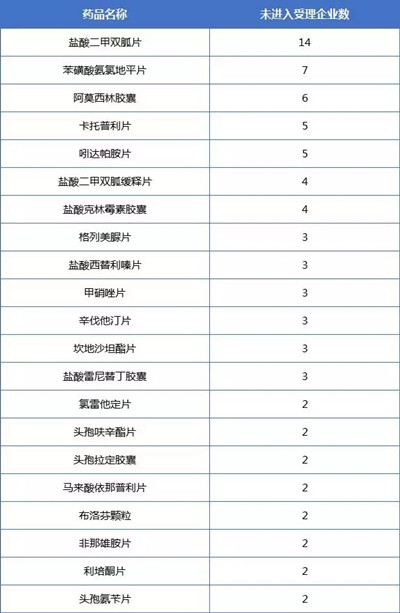
05 69个药品或将进入申报
结合目前一致性评价已进入受理的药品和企业来看,目前有142个已完成BE试验对应的药品还未进入申报;除重后可发现是来自91个企业的69种药品(分别按照企业名称和药品名称除重);下表为完成BE试验但未进入受理的品种对应的企业数排行。
总得来说,目前一致性评价相关的BE试验登记数量稳步上升,每月均有20个左右的试验终止,部分正常进入受理。
来源:药智网
政策驱动下的我国儿童药研发现状
儿童用药是近年来的热议话题,随着2013年我国人口政策的改变,单独二孩、全面二孩政策的实施,新生儿出生率及儿童占总人口的比例将大幅提升,而儿童用药安全、儿童专用药品短缺等问题一直未得到解决。近年来,政府部门为加强儿童药品的管理及可及性,出台了一系列相关政策,包括鼓励研发、优先评审、加强医院配备、招标采购直接挂网等进一步激烈儿童药的研发。本文通过梳理近几年儿童药的上市及申报信息,分析政策驱动下我国儿童用药的研发现状。
国家陆续出台一些儿童药的鼓励措施
自从世界范围内逐步重视儿童用药问题,我国也开始进入政策密集期,但2014年以前颁布的政策,并未取得良好效果,儿童用药行业依旧存在供给不足,无法满足临床需求的问题。为了进一步打消企业儿童用药的研制顾虑和推动研发机构开发临床急需的儿童适宜品种、剂型、规格的积极性,国家卫生计生委已先后发布两批鼓励研发儿童药品清单,并得到了国家重大新药创制专项的大力支持。药品监管部门也出台了加快儿童用药审评审批、成人药临床试验数据外推等一系列政策激励儿童药的开发。
表1. 儿童用药的部分政策

儿童专用药品的临床及注册申报少
通过检索CDE临床试验登记网址的数据显示,我国与儿童用药相关的临床试验登记条数仅173条记录。而通过以“儿”为关键词检索国内注册申报信息显示,2015年后申报处于仿制、进口和新药申请的受理号仅31条,而其中中药为7条,申报的药物主要以仿制药为主,较少在剂型、规格或适应症上的创新。从适应症分布来看,主要以呼吸、营养补充剂以及血液系统用药。因此,无论从儿童药剂型、适应症以及在儿童药品种的丰富上,我国在儿童药整体的研发与国外相比存在显著差距。
鼓励研发申报儿童药品种,企业研发动力不足
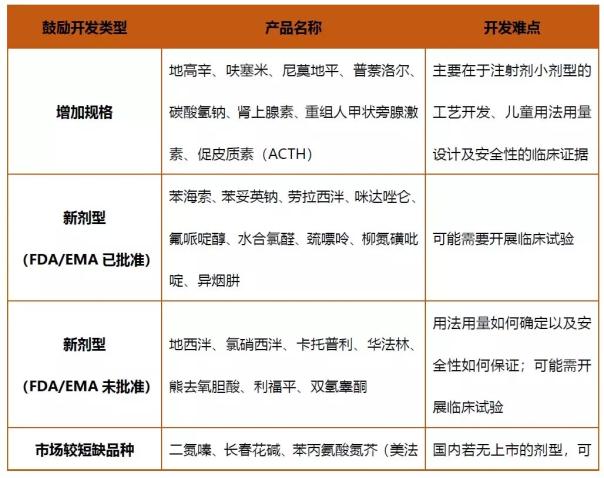
在首批鼓励研发申报儿童药品清单中,神经系统药物数量最多,占了9个名额,分别是抗抗癫痫药和精神安定药,而神经系统药物中又以精神安定药占据的比例最多。其次为血液和心血管疾病用药占6个。另外的还包括抗肿瘤、感染疾病用药、消化系统用药以及激素类产品。从治疗的适应症分布来看,主要还是用于儿童的一些难治病和罕见病的治疗。根据产品的鼓励开发类型来看,主要分为以下几类:
表2.首批鼓励研发申报儿童药鼓励开发类型
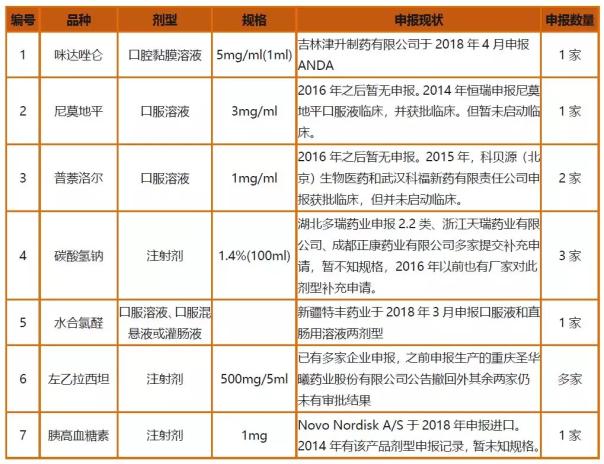
时隔两年,32个品种目前仅有7个品种有企业申报记录。清单中32种药品有25种在国内和美国均有上市;水合氯醛、双氢睾酮注射剂和凝胶2个品种3个规格在我国和美国均无上市信息;苯丙酸氮芥等4种药品仅在美国有上市药品,国内暂无已上市的同通用名药品。由此也可从侧面看到儿童药研发虽然得到政策在大力扶持,然而多数国内企业对于儿童用药的新剂型规格、罕见病治疗用药或需要开展临床的儿童药研发动力仍不足。
表3.首批鼓励研发申报儿童药品清单产品申报现状
数据来源:CDE,火石创造整理
优先审批加速儿童药上市
截止2018年9月,CDE共发布了三十二批纳入优先审评的药品清单,根据整理汇总共计575条药品信息,其中因儿童用药列入优先审评的有80条(涉及62个品种)。从2016年至2018年9月被纳入优先审评的儿童用药品种来看,近两年儿童用药申报数量有呈增长趋势,在2018年9月已达24个品种。随着政策力度的加强,未来儿童用药数量可能会出现较大幅度的增长。

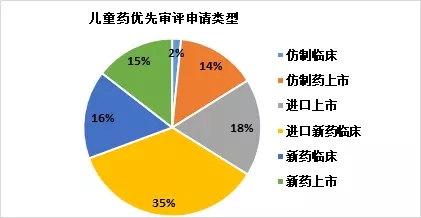
申请申报类型来看,在儿童药研发领域,外资企业表现较为积极,其中进口新药临床占据35%,进口上市占据18%;而国产儿童药在仿制药上市、新药生产和仿制临床的申请相对较少。从临床/上市的申请占比来看,其中上市申请占据了47%,优先审批在未来几年有望进一步加速儿童药上市历程。
政策利好下,儿童药龙头企业积极布局
在政策利好密集释放之下,儿童健康产业的创新发展也迎来好时代,一些儿童药的龙头企业已在积极构建儿童药研发技术平台。
康芝药业作为国内儿童健康领域的领军企业,积极构建科研创新体系,并率先成立了以儿童药为主要研究方向的企业博士后科研工作站。康芝药业于2016年获批设立海南省儿童药制剂工程技术研究中心,并有多个产品入选海南省高新技术产品。公司博士后工作站还成功组建了“口腔速溶膜剂”、“药物超微粒制备”及“药粉微观形态结构检测”三大技术平台。
葵花药业于2018年4月发布公告,公司拟1000万元投资设立全资子公司重庆小葵花儿童药物研究院,进一步加强对儿药产品的深入发掘和学术研究,提升儿童药品研发实力,巩固儿童药产品的品牌地位。
达因药业2014年,与山东大学药学院共建“儿童用药新剂型实验室”。2016年成立北京达因高科儿童药物研究院,致力于儿童高端制剂研究,打造全球领先的儿药研究平台。2017年,公司有5个儿药项目入选“十三五新药重大创制”。
小结
从现阶段的研发申报情况来看,儿童药的研发并未取得显著的效果,国内药企对儿童药的研发和申报的积极性依然不是很高。未来,政策的进一步落地,让企业实实在在享受到政策红利还需进一步推进落实。但也有业内人士也指出,解决儿童用药问题不是一朝一夕就能完成的,国家在做好顶层设计和政策支持的同时,科研院所、制药企业、临床机构要加强合作,儿药企业更要秉持匠心精神精益求精。儿童药不是最难研发的药品,但却是具有其特殊性的药品。机遇与挑战并存,对于儿童药的研发生产不仅是市场需求,为儿童的成长保驾护航也是医药社会人的责任。
来源:火石创造网
国外信息
EDQM:OMCL发布3个沙坦中NDMA检测方法
OMCLs release three methods for determination of NDMA in sartans
OMCL发布3个沙坦中NDMA检测方法
Since early July 2018 Official Medicines Control Laboratories (OMCLs) of the General European OMCL Network (GEON) have been involved in investigations and actions to address the issues related to the detection of N-nitrosodimethylamine (NDMA) in valsartan. The Network has meanwhile developed methods for the specific testing of nitrosamines in sartans on the basis different analytical principles.自2018年7月初开始,OMCL参与了调查和行动解决缬沙坦中检出NDMA事件。该网络同时开发了基于不同的分析原理的沙坦中亚硝胺物特定方法。
Three of these methods - established by the Irish OMCL inthe Public Analyst’s Laboratory in Galway (PALG), by the French OMCL at the ANSM site in Montpellier and the German OMCL at the Chemisches undVeterinär-Untersuchungsamt (CVUA) Karlsruhe on behalf of the Network - are now publicly available and can be accessed via the following links- PALG method, ANSM method and CVUA Karlsruhe method.这些方法中的3个—由爱尔兰OMCL(PALG)、法国ANSM的OMCL和德国CVUA的OMCL代表网络开发—目前发布可通过以下链接下载:
The PALG method is based on Headspace-GC-MS (single quad) and applicable to the determination of NDMA in Active Pharmaceutical Ingredients (API) and corresponding powdered tablets of the sartan group. (顶空GC-MS(单四极杆)方法,可用于检测沙坦类API及对应粉末片剂中NDMA)https://www.edqm.eu/sites/default/files/omcl-ndma-method-palg-ie-september2018.pdf
The ANSM method uses HPLC-UV as general analytical principle for the determination of NDMA in valsartan (API and drug product). (HPLC-UV方法,用于检测缬沙坦API和制剂中NDMA)https://www.edqm.eu/sites/default/files/omcl-method-determination-ndma-valsartan-ansm-september2018.pdf
The CVUA Karlsruhe method is based on APCI-UHPLC-MS/MS and applicable to the detection and quantitative determination of NDMA in valsartan drug products. (APCI-UHPLC-MS/MS方法,用于检出和定量缬沙坦制剂中NDMA)https://www.edqm.eu/sites/default/files/omcl-method-determination-ndma-valsartan-cvua-september2018.pdf
来源:蒲公英
FDA:原料药批准后变更(指南草案)
I.INTRODUCTION 概述
This guidance provides recommendations to holders of approved new drug applications(NDAs), abbreviated new drug applications (ANDAs), new animal drug applications(NADAs), and abbreviated new animal drug applications (ANADAs) and holders of drug master files (DMFs) and veterinary master files (VMFs) who want to make achange to the drug substance manufacturing process during the drug product application’s postapproval period . It does not address holders of biologics license applications (BLAs) or holders of any master files cross-referenced in BLAs.
本指南为已批准的NDA、ANDA、NADA、ANADA、DMF和VMF持有人变更其制剂声明批准之后的原料药生产工艺提供建议。本指南不适用于生物许可申报(BLA)持有人和BLA中交叉引用的任何主文件的持有人。
The guidance applies to synthetic drug substances and the synthetic steps involved in preparing semisynthetic drug substances. The guidance covers the following changes:
本指南适用于合成原料药和半合成原料药中的合成步骤。指南覆盖以下变更:
•Facility, scale, and equipment changes associated with all steps of drug substance manufacturing.
•与原料药生产所有步骤有关的设施、生产规模和设备变更
•Specification changes to starting materials, raw materials, intermediates, and the unfinished and final drug substance.
•起始物料、原料、中间体和原料药成品半成品质量标准变更
•Synthetic manufacturing process changes.
•合成生产工艺变更
•Changes in the source of the drug substance.
•原料药来源变更
•Changes to the container closure system for the drug substance.
•原料药容器密闭系统变更
This guidance does not address postapproval changes to peptides , oligonucleotides, radiopharmaceuticals; or drug substances isolated from natural sources or produced by procedures involving biotechnology; or nonsynthetic steps (such as fermentation) for semisynthetic drug substances. This guidance also does not address complex active ingredients as defined in the Generic Drug User Fee Act Reauthorization Performance Goals and Program Enhancements Fiscal Years 2018-2022, known as the GDUFA II CommitmentLetter .
本指南不针对多肽、寡核苷酸、放射药品以及从天然来源中分离的原料药和采用生物技术生产的原料药、半合成原料药的非合成步骤(例如发酵)的批准后变更。本指南亦不针对仿制药付费法案重新授权绩效目标和2018-2022财年强化计划(常称为GDUFA 承诺函)中所定义的复杂活性成分。
Ingeneral, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
一般来说,FDA的指南文件并不产生法律强制义务。相反,指南中所述的只是药监当局对某个主题当前的考虑,应仅作为是建议,其中引用了具体法规或法律要求者除外。SHOULD一词在药监机构的指南中出现时表示建议或推荐某事但并非强制要求。
II. BACKGROUND 背景
As part of the reauthorization of the Generic Drug User Fee Amendments (GDUFA II),FDA committed to issuing a guidance on postapproval changes to Type II API DMFs and submission mechanisms for ANDA holders who reference such DMFs . This guidance is intended to fulfill that commitment, and describes the recommended documentation for master file holders or drug substance manufacturers, as appropriate. The guidance also outlines the recommended documentation to be submitted by the approved application holder, as well as references the appropriate pathways for such submissions.
作为GDUFAII重新授权的一部分,FDA承诺要发布II类API DMF批准后变更以及引用此类DMF的ANDA持有人申报机制指南。本指南意在履行此承诺,阐述了建议主文件持有人和原料生产商(适当时)需提交的文件。本指南亦列出了已批准的制剂批文持有人所应提交的文件,以及对此类申报的适当引用方式。
A. Established Conditions and Reporting Categories 已订立的条件和报告分类
Under 21 CFR 314.70, 314.97, and 514.8, application holders must notify FDA about changes to conditions established in approved applications beyond the variations already provided for in their applications. FDA’s regulations identify three broad reporting categories: major changes (i.e., changes that require submission of a prior approval supplement (PAS)) ; moderate changes(i.e., changes that require submission of a changes being effected in 30 days(CBE-30) supplement or a changes being effected (CBE-0) supplement) ; and changes that must be reported in an annual report . The reporting category for a change is based on the potential risk for the change to have an adverse effect on the identity, strength, quality, purity, or potency of the drug product as these factors may relate to its safety or effectiveness. This guidance provides recommendations on the information that should be provided to CDER, CBER, or CVM to ensure continued drug substance quality and drug product quality and performance characteristics. For the most up-to-date information on reporting categories for postapproval changes, see the referenced guidances insection XII, Reporting Category.
在21CFR 314.70, 314.97和 514.8中,申报持有人必须通知FDA其对已批准申报资料中既定条件超出原有变更范畴的变更。FDA的法规大致将变更分为三种报告类别:主要变更(即需要提交预先批准的增补PAS)、中等变更(即需要提交30日内生效的增补CBE-30或立即生效的增补CBE-0),以及必须在年报中报告的变更。变更的报告类别是根据该变更对药品的鉴别、剂量、质量、纯度或效价产生的不良影响确定的,因为这些因素可能与其安全性或有效性有关。本指南对于要向CDER、CBER或CVM提交的信息提供了建议,以确保原料药质量和制剂质量和性能特性的持续性。对于批准后变更报告类型的最新更新信息,参见第XII部分“报告类别”所引用的指南。
B.Reporting Responsibilities 报告义务
Where drug substance information is provided in a DMF, a letter(s) of authorization must be provided to allow the applicant to reference the DMF . Any addition, change, or deletion of information in the master file must be submitted to the master file in the form of an amendment. Further, the master file holder must notify each person authorized to reference the DMF of the nature of the changes , and should provide as much detail as is consistent with the confidentiality agreement between the master file holder and the authorized person, so that the authorized person can determine how to report the changes in the approved application. In turn, application holders must notify FDA of each change in each condition established in an approved application, excluding the variations already provided for in the application .
如果原料药信息是在DMF里提供的,则必须提交一份授权信允许申请人引用该DMF。对DMF中信息的任何增加、变更或删除均必须以修订方式为DMF提交。另外,DMF持有人必须通知所有授权引用该DMF者其变更情况,并根据DMF持有人与被授权人之间的协议提交足够的详细信息,使得被授权人可以决定应如何在报告其已批准的申报变更。
When drug substance information is contained in an application, rather than in a referenced DMF, such changes must be submitted to FDA in the form of asupplement to the approved application or in an annual report, whichever is appropriate for the change being made .
如果原料药信息是放在制剂申报资料中而不是引用DMF,则必须采用对已批准申报资料增补的方式向FDA提交此变更,或在年报中提交(根据所做变更决定恰当方式)。
The responsibility for reporting the types of changes described in this guidance could lie with a single party or with several parties, depending on whether the drug substance synthesis or processing is described in an application or in one or more master files. The notification to FDA should include reference to the section of this guidance under which the change is made and all pertinent information to ensure the quality of the drug substance and drug product. For example, when a master file holder makes a manufacturing process change, the change should be described in an amendment to the master file, and the application holder should provide notification of the change (citing section VIII of this guidance) in a supplement or annual report, as appropriate. The data to support the process change should be provided in an amendment to the masterfile or supplement to the approved NDA or ANDA when no master file is referenced.
报告本指南中所述类别变更的义务可能是单一方的,也可能是多方的,这取决于原料药合成或加工是在制剂申报中描述还是在一个或多个DMF中描述。向FDA提交的通知应包括对本指南与所做变更相关部分的引用以及所有相关信息,以确保原料药和制剂质量。例如,当DMF持有人变更其生产工艺时,可在DMF修订中描述该变更,而制剂申报人则应在增补或年报(适当时)中提交变更通知(引用本指南第VIII部分)。支持工艺变更的数据应在DMF修订中提交,或在已批准NDA或ANDA增补中提交(如未引用DMF)。
III.GENERAL CONSIDERATIONS 一般考量
A.Assessment of Risk 风险评估
Any modification to drug substance manufacturing carries some risk of causing anadverse impact on quality, either in the physical properties of the drug substance or in the level or nature of impurities present, and, in some cases, to the bioequivalence or safety profile of the drug.
对原料药生产的任何改动均有可能对原料药质量产生不良影响的风险,要么是物理属性,要么是杂质在质与量方面,有时候是制剂的生物等效性或安全概况。
Certain kinds of modifications (e.g., equipment or facility changes) are viewed as less likely to result in an adverse impact than others (e.g., changes in the synthetic route). However, each drug substance manufacturer will need to assess the particular modification for their drug substance to determine the risk associated with the change . This guidance applies to changes made throughout the drug substance manufacturing process, i.e., from the starting material through the final drug substance. Late-stage changes in the drug substance manufacturing process are generally viewed as more likely to have an adverse impact on the quality of the drug substance and, consequently, on the drug product. Some late-stage changes should be evaluated not only by comparing pre-and post-modification drug substance, but also by comparing drug product prepared from pre- and post-modification drug substance. Finished drug product manufacturers should ensure that drug substances used in their products meet established specifications and, for compendial drug substances, meet United States Pharmacopeia (USP) standards.
某些种类的修改(例如设备或设施变更)被认为相比其它变更(例如合成路线变更)导致的不良影响会更小。但是,每个原料药生产商都需要评估其原料药的特定变更以确定该变更所带来的风险。本指南适用于对原料药整个生产工艺所做的变更,即从起始物料到原料药成品。对原料药生产工艺较后的阶段变更一般会认为更加有可能对原料药质量产生不良影响,从而对制剂产生影响。一些较后阶段的变更不仅应通过比较变更前后原料药,还应通过比较原料药变更前后的制剂来进行评估。凡是生产商应确保其制剂生产所用原料药符合既定标准,如果是药典原料药,则应符合USP标准。
Risk assessment principles are outlined in International Council for Harmonisation (ICH) guidance for industry Q9 Quality Risk Management (ICH Q9 ). As noted in ICH Q9, the level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk. A risk assessment should be performed by the drug substance manufacturer to assess the effect of the change, as well as by the drug product manufacturer to evaluate the risks associated with drug substance manufacturing modifications. A reduction in the number of drug substance and/or drug product batches from the recommendations provided in this guidance (see sections VI – XI) may also beacceptable if an adequate justification is provided based on the risk assessment.
ICH行业指南Q9“质量风险管理”(ICH Q9)中列出了风险评估原则。正如ICH Q9中所言,质量风险管理流程的工作水平、正式程度以及文件化应与风险水平相当。风险评估应由原料药生产商执行,以评估变更的影响,同时亦应由制剂生产商评估原料药生产变更所关联的风险。如果根据风险评估提交了足够的论证,则减少本指南中(参见第VI-XI部分)所建议的原料药和/或制剂批次亦可接受。
The following are examples of factors to consider when conducting a risk assessment on a change to the drug substance:
对原料药变更执行风险评估时要考虑的因素举例如下:
•Experience of the manufacturing facility and/or personnel involved in the portion of the process that encompasses the proposed change.
•所提议变更涉及工艺部分中相关生产场所和/或人员的经验
Changes implemented at the same facility with experienced personnel may pose less risk than a change implemented at a new facility with inexperienced personnel or involvement of a third-party vendor.
在相同设施内由富有经验的人员执行变更相比于在新设施由无经验的人员执行变更或涉及第三方供应商时风险会更小。
•Complexity of the manufacturing steps involved in the change.
•变更所涉及生产步骤的复杂性
Changes implemented to homogeneous reactions using common chemistry and reaction conditions may pose less risk than a change implemented to heterogeneous reaction steps involving unusually complex or sensitive chemistry, and/or unusual equipment or reaction conditions.
对采用一般化学和反应条件的均一反应执行变更相比对涉及非常规复杂或敏感化学和/或不常用设备或反应条件的成分混杂的反应执行变更风险更低
•Physical and chemical stability of the material (intermediate or drug substance) involved in the change.
•变更涉及物料(中间体或原料药)的理化稳定性
Changes implemented for a molecule that is physically and chemically stable may pose less risk than a change implemented for a molecule that degrades easily or has multiple or unstable physical forms.
对理化特性较稳定的分子执行变更相比对易于降解或具有多个或不稳定物理形态的分子执行变更风险更低
•Complexity of the molecule.
•分子复杂性
Changes implemented for a small molecule with minimal structural or stereochemical isomerism may pose less risk than a change implemented for a large molecule with multiple structural or stereochemical isomers.
对具有较少同分异构或化学立体异构体的小分子执行变更相比对具有多个同分异构或化学立体异构体的大分子执行变更风险更低
•Equivalence of the entire impurity profile.
•整体杂质概况的等同性
Batches of post-modification material that have levels of identified impurities comparable to historical data may pose less risk than batches with higher levels of identified impurities and/or new identified impurities.
变更后物料批次已知杂质水平与历史数据可比的情况相比于变更后物料批次中已知杂质和/或新已知杂质水平高于历史数据风险更低
Batches of post-modification material that have a number and level of unidentified impurities comparable to historical data may pose less risk than batches with a larger number and/or levels of unidentified impurities.
变更后物料批次中未知杂质数量和水平与历史数据可比的情况相比于变更后物料批次中未知杂质数量和水平高于历史数据风险更低
•Comparability of physical properties when they may impact drug product performance or manufacturability (typically changes made to the drug substance at or after the final solution step would bethe most likely to impact physical properties).
•可能影响制剂性能或可加工性(原料药最终溶液步骤或其后的变更一般最有可能影响物理特性)的物理特性的可比性
Post-modification material that has the same physical properties may pose less risk than post-modification material with different physical properties (e.g., solid state form, particle size, solubility,bulk/tapped density).
变更后物料具有相同物理特性的相比于变更后物料具有不同物理特性(例如,固体形态、粒径、溶解度、堆密度/击拍密度)的风险更低
B.Other Relevant Guidances 其它相关指南
This guidance aligns with existing FDA guidance, including the ICH guidances for industry Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients, Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients Questions and Answers, Q8(R2) Pharmaceutical Development, Q9 Quality Risk Management, Q10 Pharmaceutical Quality System, Q11 Development and Manufacture of Drug Substances, Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities)—Questions and Answers, and M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk.In addition to ICH Q10, which describes key systems that help establish and maintain a state of control for process performance and product quality, the FDA guidances for industry Quality Systems Approach to Pharmaceutical CGMP Regulations and Process Validation: General Principles and Practices address the current good manufacturing practice (CGMP) requirement for change control.Change control is generally understood to be the responsibility of the quality control unit . Effective change control activities are key components of any quality system. Although this guidance does not repeat the concepts and principles explained in those guidances, FDA encourages the use of modern pharmaceutical development concepts, quality risk management, and an effective pharmaceutical quality system at all stages of the manufacturing process lifecycle.
本指南与已有FDA指南保持一致,包括ICH行业指南Q7“API的GMP”,Q7问答、Q8(R2)“药物研发”、Q9“质量风险管理”、Q10“药物质量体系”、Q11“原料药开发与生产”、Q11原料药(化学实体和生物制品实体)开发与生产问答,以及M7“药物中DNA反应活性(诱变性)杂质的评估与控制以限制潜在致癌风险”。除了ICH Q10阐述关键系统用于帮助建立和维护工艺性能与产品质量受控外,FDA行业指南“药物CGMP法规的质量体系实现”和“工艺验证:通则与规范”阐述了变更控制的CGMP要求。变更控制通常被理解为质量部门的职责。有效的变更控制活动是所有质量体系的关键要素,尽管本指南并未重复这些指南中所解释的概念和原则,但FDA鼓励在生产工艺生命周期的所有阶段使用现代化药物开发概念、质量风险管理以及有效的药物质量体系。
全文参见以下链接:https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM619947.pdf
中英文网页浏览(委托发布):http://www.gjypzc.com/news?page=1&q=18
中英文PDF下载点击: FDA+原料药批准后变更+中英文+201809+.pdf
来源:蒲公英
FDA发布药品调配外包设施相关要求的指南
继2018年3月FDA发布调配药品503(B)原料药指南草案之后,2018年5月,FDA又发布了《调配药品外包设施定义指南》(Facility Definition Under Section 503B of the Federal Food, Drug, and Cosmetic Act: Guidance for Industry),对外包设施(outsourcing facilities)的定义进行了细化说明。
依据《联邦食品、药品化妆品法案》(FD&C法案)的相关规定,对调配药品的监管按照受监管主体的不同分为两类:对药房调配药品(pharmacy compounding)按照503A监管,对外包设施调配药品则按照503B监管。其主要不同之处在于,药房调配是由注册药师在州许可的药房或联邦场地(federal facility)根据患者特殊处方调配药品,可豁免cGMP要求,而外包设施则须向FDA注册登记并符合cGMP的要求。FD&C法案第503B(d)(4)条将外包设施定义为:一个地理位置或地址的设施——从事无菌药物的调配;注册登记;符合503B的所有要求。
由于某些外包设施不仅根据患者特殊处方调配药品,还可以根据其他订单调配药品,因此,FDA收到了关于外包设施定义的咨询,询问外包设施能否在其设立一个单独的区域,像药房一样在遵循503A的前提下根据患者特殊处方调配药品,而无需遵循cGMP的要求?另外,在单独的房间或邻近区域、房间调配此类药品时,是否能够使用与按照503B调配药品时相同的人员和原料?此次发布的指南重点阐释了这些问题。
根据指南中的说明,外包设施向FDA进行503B注册时,应明确其用于调配药品的所有建筑物、套间及房间。因此,在该外包设施内调配所有药品都应遵循503B并符合cGMP的要求,其中包括按照患者特殊处方而调配的药品。如果外包设施的经营者希望遵循503A调配按照患者特殊处方要求的药品,可以在独立于该外包设施以外的其他设施内设立503A调配设施,但这两个设施必须是“完全分隔”的,而不得采用在外包设施通过临时隔断(如窗帘)分隔房间或分时段调配的方式。FDA对此进行了详细的阐释和举例说明。
该指南指出,外包设施按照503A和503B不同标准调配药品时,完全分隔的目的在于避免整个生产过程的交叉和混淆,确保产品生产和质量合规,同时便于监管和检查。
原文:https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM496288.pdf
来源:CFDI网站
FDA发布两份指南为药品申请的高质量递交提供建议
美国 FDA 于 9 月 24 日发布了《ANDA 递交 — 内容和格式》定稿指南。这份长达 32 页的指南比 2014 年的指南草案更详细,提供了对文件某些部分和要求的扩展说明。同时还发布了《NDA 和 BLA 良好审评管理原则和实践》指南草案,向工业界和审评工作人员介绍了在新药审评中审评人员的职责以及良好审评管理原则和实践(GRMP)。
这两份指南的发布是 FDA 药品竞争行动计划的一部分,FDA 局长 Scott Gottlieb 表示,“作为我们不断努力降低药价和改善创新药可及性工作的一部分,FDA 发布两份指南文件协助 FDA 工作人员和行业,确保新药审评程序和仿制药审评程序管理的一致、透明和高效。”
ANDA 递交内容和格式定稿指南
ANDA 递交定稿指南旨在帮助申请人准备完整的、可递交的 ANDA 申请。定稿指南详细说明了人用药申请通用技术文件(CTD)格式中每部分的信息,并给出了 FDA 发布的支持性指南文件和建议,以帮助申请人准备其 ANDA 递交。在考虑了对 2014 年指南草案版本的公众反馈意见之后,FDA 在定稿指南中做了技术澄清并更新了参考文件。从历史上看,需要一件 ANDA 需要四轮审评周期才能获得批准,该定稿指南“通过向行业澄清如何从一开始就提交完整、高质量的申请来减少审评轮次”。
指南对 eCTD 的每部分都有建议,值得花时间仔细阅读该指南,并注意与指南草案相比的变化 , 以帮助企业更好地组织可以在“首轮”获批的 ANDA。识林将于近期推出该定稿指南与历史版本的对比解读及翻译稿。
NDA 和 BLA 良好审评管理原则和实践指南草案
这份 8 页的指南草案讨论了 FDA 审评人员在管理审评程序工作中的职责,并指出申请人如何通过良好审评管理原则和实践(GRMP)支持高效的新药或生物制品申请审评程序。指南草案表示,“GRMP 的目标是确保以一致和高效的方式管理审评程序 , 从而减少批准所需的审评周期数,并提高患者对重要治疗药的及时获取。”指南草案包含基本价值(包括问责制、沟通和一致性) , 运营原则(及时频繁的审评团队协作对于良好审评管理至关重要)以及更多关于新产品审评程序的内容。
该指南草案修订了 2005 年 4 月发布的题为《PDUFA 产品良好审评管理原则和实践》的审评人员和行业指南,这一修订工作是 FDA 作为处方药使用者付费法案(PDUFA)VI 和生物类似药使用者付费法案(BsUFA)II 的一部分承诺进行的指南更新。
来源:识林
FDA药品申请实时审评成为现实
美国 FDA 已经开始使用药品申请的实时审评,一得到试验结果就立即评估临床数据。这意味着 FDA 可以在申请人提交上市申请后立即批准新的适应症。目前,该方法仅在 FDA 肿瘤卓越中心(OCE)通过两个针对已获批抗癌药的补充适应症的试点项目实施。但之后可能会扩展到新药和新生物制剂。
在申请提交之前,FDA 就能够开始分析近期获批的 Kisqali(ribociclib)申请数据,并能够帮助指导申请人对顶线数据(top-line data)的分析,以梳理出最相关的信息。Kisqali 最初于 2017 年获批,在 2018 年 6 月 28 日提交补充申请后不到一个月的时间获得了关于乳腺癌附加适应症的批准,比目标批准日期提前数月。
根据处方药使用者付费法案(PDUFA)VI 重授权承诺函所述,药品申请的标准审评时间目标为六个月或十个月。相比之下,这两个试点项目旨在更快地批准补充申请。目标是提高抗癌药的研发和审评效率,并通过增强数据评估过程来提高 FDA 评估有效性和安全性的严格标准。
这意味着什么?对于工业界、FDA、医疗服务提供者和患者来说,这是一个双赢的局面。FDA 将有更多时间与申请人接触,并专注于药品审评的关键部分,医疗服务提供者和患者将受益于更快获得这些药品。
实时肿瘤学审评(Real-Time Oncology Review, RTOR)试点项目
FDA 正在使用的第一个试点项目称为 RTOR,重点是与评估产品安全性和有效性最相关的数据的早期提交。在信息正式提交到 FDA 之前,RTOR 允许 FDA 在临床试验结果出来且数据库被锁定之后更早地审评大部分数据。
资格标准:用于筛选符合 RTOR 的补充新药申请的标准如下:
﹒药品可能比现有治疗药物有显著改善,可能包括之前对于相同或其它适应症而获得突破性治疗认定的药品。符合其它加快审评计划(例如,快速通道、优先审评)标准的药也可以加以考虑。
﹒由审评部门和 OCE 确定的直接研究设计。仅在美国以外开展的研究以及辅助、新辅助和预防研究将被排除在外。
﹒终点易于解释(例如,随机试验中的总体存活率)。
﹒含有化学、生产和控制配方变更的补充申请以及含有药理学/毒理学数据的补充申请将被排除在外。
﹒出于试点项目的目的,也可能排除更复杂的提交,包括具有伴随诊断的提交。
流程:在得到关键试验的顶线结果时,如果符合上述资格标准,申请人可以通知相应 FDA 审评部门监管项目经理申请 RTOR 试点。审评部门和 OCE 管理层将共同决定是否可以将该申请选择加入 RTOR 试点项目。
如果 FDA 决定 RTOR 对于该申请是一个合适的审评路径,则申请人可以在输入并在数据库锁定所有患者数据且申请人准备好申请后 2-4 周内,在原始新药申请(NDA)或生物制品许可申请(BLA)下向 FDA 发送预提交数据。该数据包应包括关键原始数据集和衍生数据集,包括安全性/有效性图表、研究方案和修订,以及包装说明书草案。如适用,申请人还应提交其它学科的关键结果、分析和数据集。
FDA 将开始评估预提交数据的充分性和完整性,以解决数据质量和潜在审评问题。之后,FDA 会提供有关分析数据最有效方法的早期反馈,以正确解决关键监管问题。 当申请人向 FDA 提交申请时,审评小组已经完成了分析并熟悉了数据,可以开展更高效、及时和彻底的审评。早期参与可以提高 NDA/BLA 提交的质量以及 FDA 评估的质量。
评估协助(Assessment Aid)试点项目
第二个试点是一个新的评估协助,基于 FDA 多学科审评模板的结构化模板。多学科审评模板是申请人用于组织其提交形成结构化格式以方便 FDA 对 NDA/BLA 以及补充申请进行审评的模板。 评估协助创建了一个更高效、精简的审评流程,并通过允许 FDA 审评员专注于关键结果并执行申请人可能忽略的关键分析来减轻审评员的行政负担。反过来,评估协助带来了更加动态的审评流程,从而可以更彻底、更有效地回答关键监管问题。
流程:评估协助模板在研究用新药申请(IND)阶段发送给申请人。申请人可以在申请提交之前或在 NDA/BLA 前会议阶段通过向 FDA 监管项目经理发送兴趣通知向 CDER 血液学和肿瘤学产品办公室表明参与这一试点计划的兴趣。
一旦获得预期提交原始或补充 NDA/BLA 的顶线数据,申请人就可添加他们的立场。如果参加 RTOR 项目,申请人可以在 NDA/BLA 提交之前或前后提交评估协助。申请人部分应简明扼要,仅包括关键信息。FDA 审评团队在进行科学评估后,将使用相同的模板添加他们的评估以回应申请人的立场。FDA 的评估将关注 FDA 是否同意申请人的立场,以及任何其它发现和分析。通过使用结构化的模板,FDA 可以将其评估分层插入到申办人提交的同一文件中,允许将这一带注释的申请作为包含 FDA 审评的文件。
那些不希望参加评估协助试点的申请人将遵循通常的提交流程,不会对审评时间线或获益-风险决定产生影响。请注意 , 试点项目没有明确的时间表。在每次 CDER 血液学和肿瘤学审评部门完成试点后都将进行分析。此外,由于试点是用于补充申请的,因此没有使用者费。如果试点扩展到原始药品和生物制品 , FDA 将确定何时评定 PDUFA 费。
随着 FDA 和申请人开启这一激动人心的新探索,我们希望提高监管审评效率 , 进一步巩固我们的审评标准,同时减少可能增加审评流程时间和费用的行政负担。我们一如既往的目标是确保尽可能早的为患者提供安全、有效和高质量的治疗方案。
来源:识林