[监管动态]
我国首部化妆品安全监管地方法规出台 《广东省化妆品安全条例》7月1日施行
CDE正式发文:“特有品种”一致性评价怎么做
64个原辅包备案登记公示
国家局发布临床急需境外新药标准复核检验用资料及样品要求的通告
5化学仿制药注射剂注册生产现场检查计划下发
[行业动态]
欧盟草药科学指南系列
[国外信息]
2019年将上市的十大重磅药物
FDA挑战USP:生物制品不必符合USP标准!
新的清洁验证指南——《21世纪清洁验证》
监管动态
我国首部化妆品安全监管地方法规出台 《广东省化妆品安全条例》7月1日施行
广东省药品监管局新闻发言人、办公室主任邓林峰在举行的6月例行新闻通气会上说:《广东省化妆品安全条例》是我国第一部化妆品安全监管地方法规,广东省第十三届人民代表大会常务委员会第十一次会议于2019年3月28日审议通过,条例将于2019年7月1日起施行。
广东是占据我国化妆品产业半壁江山的大省,有化妆品生产企业2600余家,约占全国化妆品企业总数的55%。广东及时制定地方法规,有利于规范该省化妆品市场,引导企业规范生产,建立有序的市场竞争环境,全过程管控风险,促进产业发展,切实保护消费者利益。
通气会上,广东省药监局法规和科技处处长叶永才介绍说,《广东省化妆品安全条例》(以下简称《条例》)主要依据《化妆品卫生监督条例》制定,参考《化妆品卫生监督条例实施细则》等,设六章六十五条,包括总则、生产管理、经营管理、监督管理、法律责任、附则。
《条例》明确了以化妆品监管部门为主、各相关部门协同管理的部门监管职责,确定了化妆品生产经营者的主体责任。规定从事化妆品生产应当取得生产许可,产品应进行注册或备案,生产企业应加强生产管理并建立追溯制度等。
按照强化事中事后监管的理念,《条例》明确规定了经营企业主体责任,要求经营主体建立化妆品经营追溯制度。针对网络化妆品经营新业态,《条例》规定了经营者的信息告知义务和交易平台提供者对进入平台销售者的审查义务。
对于化妆品监督管理,《条例》要求监管部门对化妆品安全状况以及化妆品中的有害因素进行风险监测和风险评估,实施风险防控;监管部门有监督检查权,可采用责任约谈、暂停许可、网络证据采信等监管手段。
《条例》还规定,针对化妆品生产经营者在日常管理、标签标识管理、化妆品网络经营、化妆品委托生产等领域存在的违法行为,按照《立法法》设定的权限,设定相应的法律责任;对化妆品经营者无过错销售不符合《条例》规定的化妆品的行为,根据过罚相当原则设定免责条款;对政府监管部门不履行职责或滥用职权行为,设定相应罚则。
“《条例》实现了从化妆品卫生监督向安全监管转型,将化妆品监管重心由化妆品卫生监管延伸覆盖到化妆品卫生、质量和使用环节监管;严格化妆品生产全程控制,规定化妆品企业应当建立并执行生产管理和生产记录等制度;严格化妆品原料控制,要求用于生产化妆品所需原料应当符合国家有关标准或规定;明确化妆品标签标识规定,要求化妆品标签标注的信息以及说明应当符合国家规定,且标注的成分应当与产品实际配方一致;同时,明确了化妆品网络经营规定,要求化妆品电子商务平台应当对进入平台的化妆品经营者进行实名登记和经营资格核验。”叶永才介绍说。
据悉,目前广东省药监局已对《条例》宣传贯彻工作作出安排。《广东省化妆品安全条例释义》正在编写中,将对《条例》逐条进行解读,拟面向社会公开出版,用于宣传和培训等各项活动;该局还将制定《条例》配套制度,进一步细化、补充《条例》相关规定;同时,在全省法律骨干培训班开展《条例》培训,讲解《条例》重点和亮点。此外,广东省药监局还将组织三期面向化妆品生产企业和公众的专题培训,以普法重点班形式,对化妆品生产企业法人讲解《条例》,提高法人守法意识。
来源:中国医药报
CDE正式发文:“特有品种”一致性评价怎么做
6月21日,国家药品监督管理局药品审评中心(CDE)官网挂网《关于发布<国内特有品种评价建议>的通知》(简称《通知》),21个仿制药一致性评价“国内特有品种”名单及评价建议正式发布,《通知》旨在落实原CFDA《关于仿制药质量和疗效一致性评价工作有关事项的公告》(2017年第100号)的要求,进一步强调了企业承担的主体责任。

随着仿制药一致性评价工作深入推进,常态化、制度化的一致性评价工作已经推动产业进入高门槛的发展阶段,虽然“国内特有品种”的品种数量不多,但所包括的批准文号近2000个,涉及的企业数量高达数百家。业内更为关注的是,“国内特有品种”评价方法的确立,将进一步体现监管部门对于药品临床评价的监管思路和尺度,从而对产业链上下游带来影响。
围绕具体需求开展评价
事实上,早在2017年4月,CFDA发布的《仿制药质量和疗效一致性评价品种分类指导意见》(2017年第49号文),第六条规定:国内特有品种,由企业选择可重新开展临床试验证明其安全有效性。
2018年1月,CDE发布《关于进一步做好289基药目录中国内特有品种一致性评价工作有关事宜的通知》,发布了仿制药一致性评价289基药目录中的19个国内特有品种名单,要求企业落实评价国内特有品种的主体责任,根据品种具体情况及相关要求进行评估,提出科学合理的评价方案,并及时与CDE进行沟通。
更进一步,2018年7月,CDE发布《关于征求289基药目录中的国内特有品种评价建议的通知》,不仅将“国内特有品种”名单从19个增加至22个,更在名单基础上增加“评价建议”,配合新政策提出“无需开展临床有效性试验”“豁免生物等效性试验”“不建议企业对该品种开展再评价”等内容。
本次《通知》中所涉及的21个“国内特有品种”名单及“评价建议”,是在去年7月份二次征求意见的基础上,进一步完善和调整了名单内容,评价建议增加“本品应满足相关指导原则研究和评价要求”,对部分评价建议调整为“不推荐参比制剂”,取消了“不建议企业对该品种开展再评价”描述,并要求8个品种“完善/修订说明书”。
业内人士表示,“国内特有品种”均为上市或《药典》收载时间较早的“老药”,如盐酸小檗碱片、联苯双酯片、消旋山莨菪碱、复方利血平片等均在上世纪八十年代上市,甲状腺片、复方氢氧化铝片、复方甘草片等部分品种甚至收载于五六十年代的《药典》。
“这些品种不仅上市时间早,而且往往是价格较为便宜的产品,企业开展评价工作压力较大。”上述业内人士进一步指出,按照CDE己有的规定要求,国内特有品种的一致性评价,由企业选择可重新开展临床试验证明其安全有效性,并参照《化学药品仿制药口服固体制剂质量和疗效一致性评价申报资料要求(试行)》提交申请。
市场回报机会依然存在
仿制药一致性评价一定是常态化、制度化的举措,意味着“国内特有品种”这一特殊时期的特殊产物,将可能随着产业转型升级和去产能时代到来,得到重新的定义,“二次开发”的思路或许更适合这些特殊的产品。
行业专家表示,虽然“国内特有品种”历经临床考验,但由于历史原因,这些产品的药学研究工作在工艺、处方、标准、稳定性等方面或多或少存在一些缺陷。“遵循QbD的思路,企业需要针对工艺研究、生产、质控、风控、安全性与有效性等多个维度进行产品重塑。”
按照目前的政策要求,通过仿制药一致性评价的药品将进入《国家药品目录集》,显然,依然缺乏系统规范的资料数据的品种,即使是长期应用的经典产品,想要通过评价进入《国家药品目录集》就必须补齐这部分必要的资料数据,如药理毒理、药代、药物相互作用等方面的研究。
此次发布的《通知》,也针对部分品种提出了较为具体的评价要求。以联苯双酯片为例,围绕药物在临床上用于慢性肝炎或药物毒物引起的谷丙转氨酶(ALT)升高,但临床有效性存在争议,建议企业提供充分的临床有效性证据,临床试验终点指标不仅要纳入ALT客观指标,更应关注与损伤肝脏的治疗及保护作用相关的临床结局终点指标。
如今,国家医保局牵头的“带量采购”已经展现出一致性评价和仿制药替代的政策联动效果。行业普遍认为,只要相关品种临床疗效确切,安全性有保障,质量可控品质优良,相信通过一致性评价或者企业主动申报参比制剂之后,其投入产出一定会得到相应市场的回报。
通知原文:http://www.cde.org.cn/news.do?method=viewInfoCommon&id=314879
来源:医药经济报
64个原辅包备案登记公示
6月21日至24日,药审中心网站公示缩宫素等11个原料药登记数据、7个药用辅料登记数据和46个药包材登记数据。截至目前,药审中心已公示原料药3259个,药用辅料1658个,药包材3878个。
详细信息请见:http://www.cde.org.cn/yfb.do?method=main
来源:国家药审中心
国家局发布临床急需境外新药标准复核检验用资料及样品要求的通告
为落实《关于临床急需境外新药审评审批相关事宜的公告》(2018年第79号)要求,加快临床急需境外上市新药审评审批,我局组织制定了《临床急需境外新药标准复核检验用资料及样品要求(化学药品)》和《临床急需境外新药标准复核检验用资料及样品要求(生物制品)》(见附件),现予发布。凡列入国家药监局药品审评中心公布的《临床急需境外新药名单》的品种,申请人应当在申报药品上市时按照本通告要求同步向中国食品药品检定研究院提交用于药品标准复核检验的相关资料及样品。
本通告自发布之日起实施。
原文:http://www.nmpa.gov.cn/WS04/CL2138/338476.html
来源:国家药监局
5化学仿制药注射剂注册生产现场检查计划下发
国家药品审核查验中心发布《关于注射用头孢美唑钠等5个化学仿制药注射剂注册生产现场检查计划的通告》对不同生产企业的头孢美唑钠等5个化学仿制药,进行现场检查。
原文:http://www.cfdi.org.cn/resource/news/11566.html
来源:国家药品审核查验中心
行业动态
欧盟草药科学指南系列
草药制剂的生产
由于草药药品的复杂性和多样性,对起始原料的控制、贮存和加工在草药药品制造中尤为重要。
草药药品制造中的“起始物料”可以是药用植物、药材或草药加工品。药材应有合适的质量,质量相关支持性数据应当提供给草药加工品/草药药品制造企业。为保证药材质量的一致性,可能会对农业生产方面的信息要求得更为详细。选种与收获条件代表着药材质量的重要方面,并可能影响成品的一致性。
厂房与设备
贮存区域
药材应贮存在单独区域。贮存区应配有防虫或防止其他动物进入的设备设施,特别是啮齿类动物。应采取有效措施防止动物和微生物在药材上的传播,防止其发酵、霉变及交叉污染等。应使用不同的隔离区域来区分入厂待验的药材与合格的药材。
贮存区域应该通风良好,且容器的放置方式应允许空气自由流通。应特别注意贮存区域的清洁与维护,尤其是易产生粉尘的地方。药材与药草加工品的贮存可能对湿度、温度和光线具有特殊要求,应对贮存条件进行监控。
生产区域
药材与草药加工品的取样、称量、混合与加工等过程,一旦产生粉尘,应有特殊规定,以便于清洁和防止交叉污染。例如使用除尘系统、专用厂房等。
设备
制造工艺所用的设备、过滤材料等必须与提取溶剂相适应,以防止释放或吸附任何可能影响产品质量的物质。
文件管理
起始物料的质量标准
草药药品制造企业必须确保仅使用按GMP与上市许可注册要求制造的草药起始物料。由草药药品制造企业或其代表对供应商的全面审计资料应当可用。原料药的“审计追踪”对起始物料的质量至关重要。制造企业应确保药材/草药加工品的供应商符合《植物源起始物料种植和采集质量管理规范》要求。
为了满足《欧盟GMP指南》基本要求所描述的质量标准要求,药材/草药加工品的文件管理应当包括以下内容:
植物双学名(属、种、亚种/变种与发现者)。如可能,也应提供其他相关信息,例如栽培种名称与化学型;植物来源详细情况,如原产地、种植、收获时间、采集规程、可能使用的杀虫剂、可能的放射性污染等。
使用植物的哪些部分。如果使用干燥植物,应说明所使用的干燥系统,对药材的性状及宏观和微观检查等。
合适的鉴别。如果适用的话,包括对已知的治疗活性成分或标记成分做鉴别。如果药材有被掺假/替换的可能,则要求有特异性强的鉴别(以区别掺假物或替代物)。应具备用于鉴别参考的真实样本。
按照《欧洲药典》要求测定的药材的含水量。对已知药理活性成分或对标记成分(如果适用的话)进行含量分析。除非另有合理性论述,含量分析方法应适于可能的农药污染,并含有可接受限度,符合《欧洲药典》要求,如果《欧洲药典》没有规定,应使用经过恰当验证的方法。
适当检测真菌和(或)微生物污染,包括黄曲霉素、其他真菌毒素、虫害,并含有可接受限度。
适当检查重金属及类似的污染物和掺假物。适当检查异物。
按照《欧洲药典》关于药材总论或具体药材各论要求,附加适当的任何其他检测。任何用来降低真菌/微生物污染或其他虫害的处理均应文件化。应当有质量标准和规程,并包括详细的过程、检测和残留限度。
加工操作法
加工操作应描述药材的不同操作,如清洗、干燥、粉碎、过筛,包括干燥时间和温度,以及用于控制碎片或颗粒大小的方法。
须注意的是,应有书面操作法和记录,来确保每个容器中的药材经过细致检查,以检测是否存在掺假/替换,或存在异物,例如金属或玻璃屑、动物残骸或粪便、石头、沙子等,或腐烂与变质迹象。
加工操作法也应描述安全筛分或除去异物的方法,以及在贮存合格的草本物质前或开始生产前,所需的植物物料清洗/挑选的适当规程。
对于草药加工品的生产来说,操作法应包括提取用溶剂、提取时间与温度的详细内容,以及浓缩步骤和使用方法的详细内容。
质量控制
样品
由于药用植物/药材本质的多样化,应当由专业人员谨慎取样。每批都应当使用专属于该批的文件进行鉴别。
必须有植物物料的对照品,特别是在药材没有收载到《欧洲药典》或其他成员国药典的情况下,如果使用粉末,则需要未粉碎的植物物料样品。
质量控制人员应当具有关于药材、草药加工品和(或)草药药品方面的专业知识和经验,以便能够进行鉴别试验及识别掺假、真菌生长、虫害感染、同批交货的粗品物料不均匀等问题。
药材、草药加工品与草药药品的鉴别与质量检验,应当按照相关现行的传统草药药品和草药药品质量标准欧盟指南执行,如果相关,按照《欧洲药典》特定的各论执行。
草药/传统草药药品稳定性检测
草药/传统草药药品的质量,包括稳定性,应予以保障并符合相关要求。具体要求在修订的2001/83/EC 法令附录1、2001/82/EC 法令附录1 以及现行的EU/ICH 质量指南中陈述。欧洲药品管理局有关委员会已发布了有关稳定性检测方面的一些指南,这些指南主要集中在化学定义物质方面。鉴于草药药品复杂的自然属性,欧洲药品管理局有关委员会考虑出台进一步的指南,以保证并强调这些产品的稳定性。
问题陈述
相比化学定义物质,评估草药药品的稳定性挑战较大,尤其在以下几方面:
草药药品中的活性物质[药材和(或)草药加工品]包含复杂的成分,而且在许多情况下具有治疗效果的成分是未知的。当某个药品中含有两个或两个以上的药材和(或)草药加工品时情况更加复杂。在许多情况下,草药药品为药材和(或)草药加工品复合物时,由于它们拥有相似成分,增加了分析的难度。
考虑到草药药品具有的特点,目前已建立了适当的质量概念。作为药材、草药加工品和草药药品整个控制策略的一部分,一系列检测标准,包含定性和定量参数已被认可作为质量指标。
关于稳定性测试,指纹图谱以及通过药材标记物建立的含量测定方法已在储存期质量标准中写明。尽管这种方法有可行性,在实际情况中通常会随之带来分析方面的问题和较高的成本。
总而言之,草药药品具有许多特性,这些特性明显不同于传统定义的药品,因此需要建立特殊的稳定性指导原则,这些原则应覆盖现行的草药药品特殊要求和稳定性通用指导原则中未强调之处。
结论
应强调在草药药品行业中建立稳定性指南原则的重要性,同样重要的还有何时应用简化的稳定性测试。为帮助申请者选择合适的稳定性方案,监管部门应频繁提及关于这方面的问题。这些问题主要涉及传统草药药品注册方面,这些药品通常包含许多活性物质。尽管有许多情况需要加以具体特殊强调,一些案例也许有通用之处,或可提供关于草药药品稳定性通用指导的基础依据。
熏蒸剂的应用
草药药品的质量取决于起始植物物料的质量、中间过程控制、GMP 控制、过程验证,以及贯穿于开发和生产所应用的技术参数等。
植物来源的产品质量一致性只能基于在对植物物料严格和详细定义下予以保证,尤其是需要对所使用的植物物料进行明确的植物学确认。重要的是,需要明确地域来源和药材收获的条件,以便能够保证物料持续的质量状态。另外,根据欧洲药品法案,草药质量档案应强调潜在的污染,如微生物、微生物类产品、杀虫剂、有毒金属、放射性污染、熏蒸剂等。因此,应对熏蒸剂潜在的残留进行充分评估。
问题陈述
自1989年12月31日起,欧洲禁止使用环氧乙烷熏蒸药材。另外,根据1992年的蒙特利尔议定书,由于生产企业和申请者常用的熏蒸剂溴甲烷是一种消耗臭氧的物质,因此它也逐渐在世界范围内退出。此外,对于草药药品所使用的药材,生产企业和申请者需要考虑其他控制虫害的策略。
结论和建议
熏蒸剂的效力只能通过对植物物料、库房和生产厂房正确的管理加以实现和维持。应基于对所保护的植物物料风险评估来采取相应的策略,这需要适当的预防、监测和虫害控制等方法。
在选择其他策略时,应考虑以下几个方面:
整体策略的效果。包括使用熏蒸剂的必要性,熏蒸剂的选择和采用的熏蒸时间,对所采用的熏蒸方法在物理化学、毒理学、生态环境特性等方面进行风险评估,尽可能减少对工人、消费者、环境和药材本身的风险,应对熏蒸剂残留采用适当的检测、限度、分析方法。
总之,建议应尽可能限制熏蒸剂的使用。熏蒸剂应该只有在被证实确实需要时才能使用,并应由经培训的合适员工结合所使用熏蒸剂的特殊建议进行处理。
来源:以上文章均摘编自《欧盟草药科学指南》
国外动态
2019年将上市的十大重磅药物
FiercePharma发布《2019年将上市的重磅药物TOP10》。该榜单基于EvaluatePharma在今年1月和2月发布的预测报告,这些药物将在2019年上市、并将在2024年实现重磅销售。值得注意的是,生物制药行业最受欢迎的癌症适应症,在这份榜单中消失了;相反免疫学和罕见基因疾病成为了焦点。
具体排名如下:
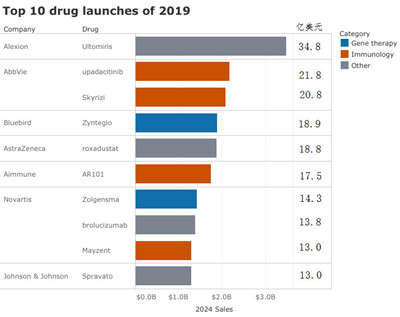
榜单显示,Alexion公司Soliris的升级版Ultomiris将成为今年推出的最具商业潜力的新药,该药于2018年12月获得美国FDA批准,比预期提前了两个月,预计2024年销售额将达到34.8亿美元。
目前,Alexion正在调整战略重点,由“超罕见”转向“罕见”疾病,以接触更多的患者群体。作为计划的一部分,公司已将Ultomiris定价低于Soliris的10%,并计划将Soliris的70%患者转向Ultomiris,同时该药是目前唯一一个每8周给药一次的长效C5补体抑制剂,已获批的适应症为阵发性夜间血红蛋白尿(PNH)。就在最近,Ultomiris治疗非典型溶血性尿毒综合征(aHUS)的新适应症申请获得了FDA的优先审查,预计今年10月中旬做出批准决定。
榜单中有两款基因疗法,分别为诺华脊髓性肌萎缩症药物Zolgensma和蓝鸟生物β地中海贫血药物Zynteglo。基因疗法近年来引起了行业的浓厚兴趣,同时也引发了许多争议。作为一次性治疗药物,这类疗法具有长期疗效承诺,但定价一直受到各界密切关注,不过上述两家公司都提供了新的治疗模式,如多年分期付款。
值得注意的是,诺华在榜单中拥有3款药物,在所有公司中具有最强的竞争力。除了Zolgensma,另两款药物是眼科药物brolucizumab和多发性硬化症药物Mayzent。Brolucizumab是一款人源化抗体单链可变区片段(scFv),独特的创新结构使其仅有26kDa大小,针对所有亚型VEGF-A具有强烈抑制作用,目前治疗新生血管年龄相关性黄斑变性(nAMD)正在接受FDA的优先审查。上市后,brolucizumab将与拜耳/再生元的Eylea及诺华/罗氏合作开发的Lucentis展开竞争。头对头临床研究中,在部分次要终点方面,brolucizumab疗效优于Eylea。
Mayzent则是首个治疗继发进展型多发性硬化症(SPMS)的口服药物,于今年3月获得FDA批准。据估计,高达80%的复发-缓解型多发性硬化症患者会发展为SPMS,但挑战在于如何帮助医生更有效地识别这些患者。目前,诺华正致力于医师和患者教育,帮助提高对这一阶段疾病及其症状的认识。
艾伯维有两款新产品上榜,分别为Skyrizi和upadacitinib。目前,该公司正试图使其投资组合多样化,应对Humira生物仿制药带来的竞争。瑞士信贷分析师在最近的一份报告中预测,无论是关节炎、哮喘、炎症性肠病还是多发性硬化症,这些抗炎领域的每个分市场都有着数十亿美元的增长空间。
榜单中,还包括来自Aimmune Therapeutics公司的花生过敏免疫疗法AR101,该药的目标是成为第一种可预防儿童意外摄入花生导致过敏免疫反应的药物,其上市申请在去年12月提交,但不幸遭遇美国政府停摆被推迟审查,目前正在与FDA商讨加速审批。
榜单中有两款药物在上市过程中可能面临新问题。一是贫血药物roxadustat,该药由FibroGen与阿斯利康和安斯泰来合作开发,已于去年12月在中国获得全球首批。然而,最近针对慢性肾脏病患者III期临床研究的一项安全性汇总分析却发现了一些令人困惑的安全信号。二是强生的抗抑郁症药Spravato鼻腔喷雾剂,该药是30年来首个具有新作用机制的重度抑郁症药物,但无论是价格标签还是安全风险都存在争议。
在发布TOP10榜单时,蓝鸟生物披露Zynteglo的商业发射将延迟,但该药仍被列入了榜单,原因是蓝鸟生物已计划在今年内招募患者并在2020年初启动首批患者的商业化治疗。
FiercePharma指出,今年的榜单显示了基于新技术的昂贵罕见病药物的蓬勃发展,以及对不断增长的免疫市场的持续热情。
来源:The top 10 drug launches of 2019/新浪医药新闻
FDA挑战USP:生物制品不必符合USP标准!
近日,美国药典委员会正在反对一项法案,该法案规定生物制品不必符合 USP 质量标准要求。
FDA 药品审评与研究中心(CDER)主任 Janet Woodcock 以及生物制品审评与研究中心(CBER)主任 Peter Marks 在 6 月 13 日的一篇博客文章中表示,当涉及生物制品(包括生物类似药)时,要求遵照USP专论实际上可能阻碍发展,而不会提供额外的质量保证。”
他们表示,FDA 已经制定了确保生物制品(包括生物类似药和可互换产品)的安全性、纯度和含量(安全性和有效性)的标准。要求遵守非政府机构(美国药典委员会是世界上唯一一家非政府的药典机构)发布的专论可能会迫使申办人在只需要遵守一个标准的情况下,不得不同时遵守两个标准,这可能延长审评时间,并延迟批准所需的时间,从而减少竞争并限制创新。
Kozlowski 最后强调了 FDA 对小分子药品专论标准的持续支持。他还表示支持符合 FDA 灵活方法的生物制品可选标准,“但任何强制性标准都会阻碍创新技术,并给行业和审评人员带来不必要的负担。”
FDA Challenges USP Standards for Biologics
FDA 挑战 USP 生物制品标准
A provision in a Senate health reform bill has reignited debate over the whether biological products should have to meet product quality standards established by the U.S. Pharmacopeia. FDA officials contend that efforts by the USP to publish product-specific monographs for biological products could hinder the development of innovative products, including competitive biosimilars. USP officials, with support from pharmacists and some patient and health care groups, maintain that monograph standards are important for ensuring the safety and quality of all drugs and biotech therapies.
参议院医疗改革法案中的一项条款重新引发了关于生物制品是否必须符合美国药典规定的产品质量标准的辩论。FDA官员认为USP生物制品专论可能会阻碍创新产品的开发,包括有竞争力的生物仿制药。而USP官员在药剂师以及一些患者和医疗团体的支持下,坚持认为专论标准对于确保所有药物和生物技术疗法的安全性和质量非常重要。
The new policy sought by FDA is contained in bipartisan legislation recently issued by the Senate Health, Education, Labor and Pensions (HELP) committee, which presents multiple provisions designed to improve national health care programs and access to medicines. These include items to revise patent policy, generic drug testing, and biosimilar oversight as part of efforts to reduce the cost of drugs and biologics.
FDA的新政策包含在参议院卫生、教育、劳工和养老金委员会(HELP)最近颁布的两党立法中,其中提出了多项条款,旨在改善国家医疗保健计划和获得药品的机会。.这些项目包括修订专利政策、仿制药测试和生物仿制药监督,作为降低药物和生物制剂成本工作的一部分。
The disputed language (section 207 of S. 1895) excludes all biological products regulated by the Public Health Service Act from meeting USP compendial standards. In a recent posting on the FDA website, Janet Woodcock, director of the Center for Drug Evaluation and Research (CDER), and Peter Marks, director of the Center for Biologics Evaluation and Research (CBER), warn that continued innovation in this area could be blocked by “inflexible standards that can’t evolve quickly enough to keep up with technological development,” as might occur if product-specific USP standards are applied to biologics. FDA already has standards in place to ensure the safety, purity and potency of biological products, Woodcock and Marks observe, and the need to comply with additional standards could extend reviews and delay approvals.
该有争议的文字(S.1895第207条)排除了《公共卫生服务法》规定的所有生物制品必须符合USP标准。在FDA网站上最近发布的文章中,药物评价和研究中心主任珍妮特•伍德科克和生物制剂评价与研究中心主任彼得•马克斯警告说,这一领域的持续创新可能会受到"不灵活标准的阻碍,这些标准发展速度不够快,无法跟上技术发展",如果特定产品的USP标准应用于生物制剂,就可能发生这种情况。FDA已经制定了标准以确保生物制品的安全性,纯度和效力,而如果还要符合额外的标准则可能会扩大审查范围并推迟审批,伍德科克和马克斯说。
Similarly, Steven Kozlowski, director of CDER’s Office of Biotechnology Products, explains in another commentary that most widely used biological products do not have USP monographs, and that efforts to establish mandatory standards for these inherently complex and variable products could “impede technological progress or innovation.” Kozlowski raises the possibility that the manufacturer of an older innovator therapy could ask USP to write a monograph for its product citing patented characteristics that would produce “unnecessary barriers to innovation and progress.” Unlike small molecule drugs, which are chemically synthesized to achieve “sameness” with the innovator drug, biological products are highly complex proteins produced from living cells and aim to be “highly similar,” but not the same, as the reference product.
同样,CDER生物技术产品办公室主任史蒂文•科兹洛夫斯基在另一篇评论中解释说,大多数广泛使用的生物制品没有USP专论,而为这些复杂、多变的生物制品制定强制性标准可能会"阻碍技术进步或创新"。Kozlowski 提出,一种较旧的创新疗法的制造商可能会要求 USP 为其产品撰写专论,写入独家的特性,这些特性将"对创新和进步产生不必要的障碍"。
Kozlowski maintains that adhering to USP standards would not necessarily prevent products from experiencing quality or safety problems and could make it difficult for a subsequent manufacturer to implement changes, such as a more efficient manufacturing process. While FDA supports USP monograph standards for conventional drugs, it prefers optional standards for biological products that align with the agency’s flexible approach for inherently complex biological products.
Kozlowski 认为,符合USP 标准并不一定能防止产品出现质量或安全问题,但可能使后续制造商难以实施变更,例如实现更高效的生产过程。虽然FDA支持USP常规药物的专论标准,但它更喜欢生物制品的可选标准,这些标准与FDA对固有复杂生物产品的灵活方法保持一致。
USP fights back
USP的回应
A coalition of pharmacists, public health and patient organizations supports efforts by USP to convince the authors of the Senate HELP bill to drop the curb on compendial standards for biologics. The group maintains that the proposed policy change would weaken assurances that medicines are safe and of high quality, without doing anything to lower drug costs. USP officials note that mandatory drug quality standards developed by the European Pharmacopeia and the World Health Organization have supported drug development and approval, including biosimilars, in Europe and other regions.
一个由药剂师、公共卫生和患者组织组成的联盟支持USP工作说服参议院HELP法案的作者放弃对生物制剂的合成标准的要求。该小组认为,拟议的政策变化将削弱药品安全和高质量保证,而不会降低药物成本。USP官员指出,欧洲药典和世界卫生组织制定的强制性药物质量标准支持了欧洲和其他地区的药物开发和批准,包括生物仿制药。
来源:GMP办公室
新的清洁验证指南——《21世纪清洁验证》
Pharmaceutical Online发布了关于清洁验证的新指南——《21世纪清洁验证》,全文100页,包含以下8部分内容:

1、开发基于科学、风险和统计方法的清洁工艺和验证
2、使用ADE评估共享设施产品交叉污染风险
3、过程能力方法用于评估共用设施交叉污染风险
4、擦拭法用于评估TOC可检测性
5、用于评估目视检查可检测性方法
6、如何验证目视检验作为清洁验证的分析方法
7、目视检查用于低风险,多产品共用设施清洁验证的论证和确认
8、清洁风险测量和清洁风险列表
解读如下:
﹒给出了清洁工艺开发和验证过程的风险管理活动,包括如下:
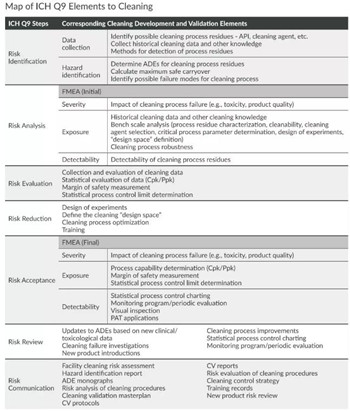
﹒就FDA《工艺验证指南:一般原则与规范》如何应用于清洁工艺开发和验证提供指导:
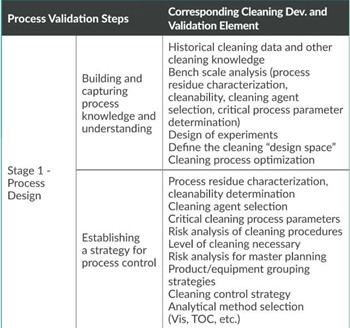
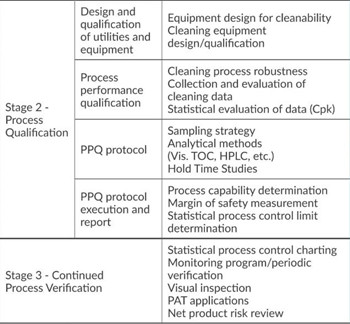
﹒对于交叉污染风险的评分,给出了一种基于ADE值的评分标准,例如ADE为1g/天时,对应的风险赋分为0;相反,ADE为100pg/天,则风险赋分为10.
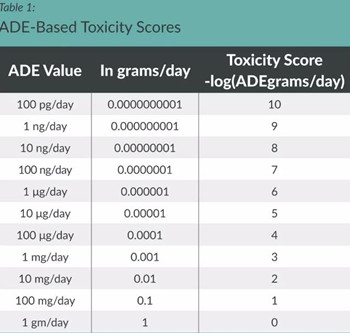
﹒文章给出一个例子,4个车间A、B、C、D分别生产5种产品,并具备不同的ADE风险赋分:
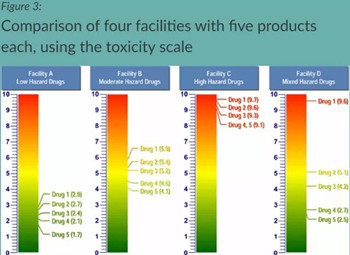
﹒对于验证策略而言,A车间可能不需要采取与C车间相同的验证程序,如果计算出的表面最大允许残留(MSSR)远远高于可见水平,并且所有表面都能够被检查,那么实际上可以使用“目视检查”来作为其清洁验证的唯一接受标准;而C车间的MSSR可能低于可见水平,需要进行擦拭/冲淋取样,甚至可能需要特定的分析方法,可能需要持续进行监测;D车间具备低、中、高毒性药品共线,这种情况下,可能需要在药品1(毒性评分=9.6)之后进行全面的生产控制、擦拭/冲洗、取样和持续监测。但是该车间的药物4(毒性评分=2.7)和药物5(毒性评分=2.5)则可能只需要进行“目视检查”作为清洁验证的唯一接受标准即可。
﹒给出应用过程能力指数(CPK)评价清洁验证的方法
﹒给出根据清洁工艺过程能力指数(CPu)对交叉污染风险可能性的评分表格:
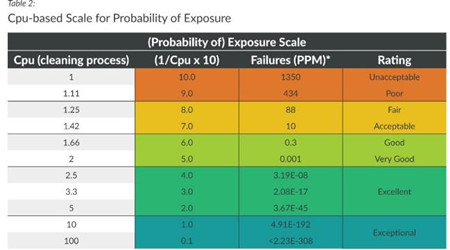
﹒文章认为在评估交叉污染风险时,直接严重性*可能性并不合适,文章引入了SO(风险评分)的概念,仍然保留基础信息。
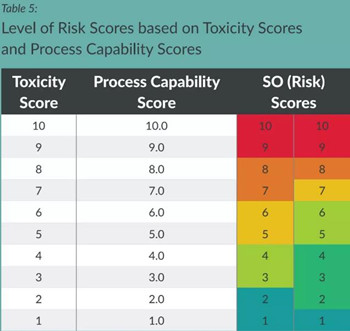
﹒例如,10/10"意味着非常有毒的化合物,而清洁能力非常差,这意味着非常危险的情况。“10/1"意味着非常有毒的化合物,但清洁是非常有效的,这将导致非常低的风险情况。“5/5"意味着这是 种中等毒性的化合物 ,而且清洁效果很好,所以风险很低。如果是“5/10”,那就意味着这是一种中等毒性的混合物,而且清洁效果很差,风险很高。“5/1”意味着它也是种中等毒性的化合物,但它的清洁效果非常好,因此风险非常低。
﹒文章给出了TOC方法用于检测清洁验证残留的论证和分析方法验证要求

﹒文章指出,最近,监管机构对目视检查作为清洁验证唯一接受标准有所放开,并给出目视检查检出限的确认方法
﹒文章给出交叉污染FMEA风险评估严重性、可能性和可检测性的评分细则

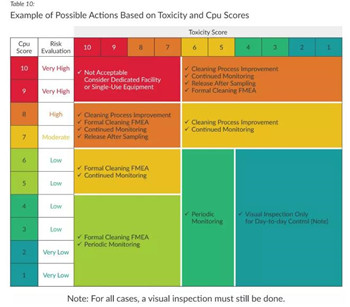
﹒根据风险评估结果,对于不同的毒性和CPu赋分组合,给出不同的风险控制措施,例如,毒性赋分=10且CPu赋分=10的情况是不可接受的,需要考虑专用设施或一次性使用设备进行生产;毒性赋分=1且CPu赋分=1则仅需要在日常清洁后进行目视检查即可。
原文下载:Part 1:开发基于科学、风险和统计方法的清洁工艺和验证.pdf、Part 2:使用ADE评估共享设施产品交叉污染风险.pdf、Part 3:过程能力方法用于评估共用设施交叉污染风险.pdf、Part 4:擦拭法用于评估TOC可检测性.pdf、Part 5:用于评估目视检查可检测性方法.pdf、Part 6:如何验证目视检验作为清洁验证的分析方法.pdf、Part 7:目视检查用于低风险,多产品共用设施清洁验证的论证和确认.pdf、Part 8:清洁风险测量和清洁风险列表.pdf
来源:GMP办公室