本 期 要 目
[焦点关注]
《2018年度药品审评报告》发布
《2018年度药品审评报告》五大看点解析
《疫苗管理法》正式公布 将于今年十二月一日起施行
[监管动态]
CDE发布中医药产业3项技术指导原则
国家药监局:医疗器械唯一标识系统试点工作方案
[综合分析]
2019年6月CDE药品审评情况报告
一致性评价最新进展:受理号达1197个,通过216个,视同通过77个
盘点:2019年上半年,中国1类新药IND数量57个
[国外信息]
盘点:2019年1-6月,FDA批准的8个NME!
盘点2019年上半年获批新药中的“首例”
盘点7月将面临美国FDA重要监管决定的5个新药
欧盟《生产商符合欧盟防伪法令的GMP检查备忘录》(中英文)
欧盟发布《序列化追溯系统GMP检查指南》
欧盟/美国GMP互认:德国最终被确认为第27个国家!
焦点关注
《2018年度药品审评报告》发布
国家药监局重磅发布《2018年度药品审评报告》,内容包括:
﹒药品注册申请受理情况(共7336件)
﹒药品注册申请审评审批情况(共9796件)
﹒鼓励创新与保障公众用药情况
﹒主要工作措施及进展情况
﹒2019年重点工作安排
﹒药审中心审评通过的1类创新药
﹒药审中心审评通过的进口原研药
﹒2018年审评通过的优先审评药品名单
﹒2018年通过一致性评价的品种
﹒第一批临床急需境外新药的审评审批情况
﹒2018年药审中心起草经国家局发布的技术指导原则
详细信息请见:http://www.nmpa.gov.cn/WS04/CL2196/338621.html
来源:国家药监局
《2018年度药品审评报告》五大看点解析
7月1 日,国家药品监督管理局药品审评中心发布《2018年度药品审评报告》(以下简称《报告》)。《报告》显示,2018年我国药品审评审批改革成效显著,新药上市跑出了加速度,人民群众的用药需求得到了更好保障。药审中心全年审评通过106个新药(按品种统计),包含2个新中药复方制剂,以及9个1类创新药和67个进口原研药;313件注册申请被纳入优先审评程序,其中83个药品通过优先审评程序得以加快批准上市;57个品种通过口服固体制剂一致性评价;第一批遴选的48个临床急需境外新药中,10个品种获批上市。
持续深化药审改革
为更好地满足临床需求,国家药监部门持续深化药品审评审批制度改革,确保各项改革任务落到实处。
2018年,药审中心对2007年以来在美国、欧盟或日本批准上市,但尚未在我国境内上市的新药进行梳理,组织专家遴选出第一批48个临床急需新药,纳入专门通道加快审评,多措并举加大对申请人的服务和指导。48个境外新药中,已受理17个品种,10个品种已获批上市,7个正在进行技术审评。
为使更多新药能够及早进入临床试验,药审中心制定了《临床试验默示许可审评审批工作程序》,实施临床试验默示许可制,大幅减少临床试验审评用时,在保证审评质量的同时提升审评速度。
同时,药审中心还进一步深化、细化、实化适应症团队、优先审评、沟通交流、立案审查制度等工作,实行原辅包与制剂共同审评审批,运行上市药品目录集系统,改革临床试验管理,探索开展合规工作。
注册受理增长47%
《报告》显示,2018年,药审中心受理新注册申请共7336件,其中需技术审评的注册申请5574件,直接行政审批(无需技术审评)的注册申请1762件。需技术审评的注册申请任务受理量较2017年增长了47%,且中药、化药和生物制品各类药品注册申请任务受理量均有较大幅度增长。
但注册申请审评用时明显下降。至2018年底,药审中心实现中药、化药、生物制品各类注册申请按时限审评审批率已超过90%。全年完成审评审批的注册申请共9796件,其中完成需技术审评的注册申请7988件;无需技术审评的直接行政审批任务1808件,平均审批时限为12.3个工作日,远小于法定的20日行政审批时限。
排队等待审评审批的注册申请已由2015年9月高峰时的近22000件降至3440件,进一步巩固了解决注册申请积压的成效,基本完成了国务院《关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)确定的2018年底实现按规定时限审评的工作目标。
研发创新态势明显
受益于药审改革加速,2018年创新药注册申请数量大幅增长,药品研发创新态势明显。《报告》显示,药审中心2018全年受理1类创新药注册申请264个品种(化药的品种数以活性成分统计,共157个;中药和生物制品的品种数均以药品通用名称统计,中药1个,生物制品为106个),较2017年增长了21%。其中,1类创新药的新药临床试验(IND)申请有239个品种,较2017年增长了15%;1类创新药的新药上市申请(NDA)有25个品种,包括16个化学药品、9个生物制品,较2017年增长了150%。
值得注意的是,在1类创新药注册申请的264个品种中,有222个品种来自国内企业,适应症主要集中在抗肿瘤、内分泌系统和消化系统领域;42个品种为进口药,适应症主要集中在抗肿瘤、循环系统和消化系统领域。
新药上市进程加快
通过实施优先审评程序、实施临床试验默示许可制、加强与申请人的沟通交流等措施,药审中心在2018年审评通过一批具有明显临床价值、临床急需等新药好药,建议批准上市,为患者提供了更多用药选择。
《报告》显示,药审中心全年审评通过新药(NDA及IND直接批产)106个,包含关黄母颗粒、金蓉颗粒2个新中药复方制剂,以及9个1类创新药和67个进口原研药。9个1类创新药全部为我国自主创新药品且以抗癌药、抗病毒药居多,分别是呋喹替尼胶囊、罗沙司他胶囊、马来酸吡咯替尼片、盐酸安罗替尼胶囊、达诺瑞韦钠片、注射用艾博韦泰6个化学新分子实体药物,特瑞普利单抗注射液、信迪利单抗注射液2个抗PD-1单克隆抗体,以及重组细胞因子基因衍生蛋白注射液。
根据《关于鼓励药品创新实行优先审评审批的意见》,2018年有313件注册申请被纳入优先审评程序,其中23%为具有明显临床价值的新药注册申请,儿童用药和罕见病用药有63件。83个品种通过优先审评程序得以加快批准上市(以通用名计算)。
为提升我国仿制药质量,药审中心全力推进仿制药质量和疗效一致性评价(以下简称一致性评价)工作,2018年全年正式发布了8批参比制剂目录(221个品规),受理口服固体制剂一致性评价申请440件(155个品种),通过111件(57个品种),其中289基药品种申请63件(36个品种)。
依法履职科学审评
在持续深化药品审评审批制度改革的同时,药审中心坚持依法依规、科学规范审评,进一步加强审评科学基础建设。强化审评质量管理,并加强对自由裁量权的制约和监督。
2018年,药审中心起草经国家局发布《新药I期临床试验申请技术指南》《创新药(化学药)Ⅲ期临床试验药学研究信息指南》《证候类中药新药临床研究技术指导原则》等指导原则17个 。
为进一步方便企业研发申请,药审中心不断丰富沟通渠道,提高沟通效率和质量,形成了沟通交流会议、网络平台咨询(一般性技术问题)、电话咨询、邮件咨询和现场咨询的多渠道、多层次的沟通交流模式。2018年,药审中心接收沟通交流申请1982件,较2017年的840件增长了136%;接收网络平台咨询15219个,较2017年的5881个增长了159%,同时每周三定期开展现场咨询。
2018年6月,国家药监局当选为国际人用药品注册技术协调会(ICH)管理委员会成员。药审中心在网站上开设了“ICH工作办公室专栏”,以便业界及时了解ICH工作动态,积极参与ICH相关工作。
来源:中国食品药品网
《疫苗管理法》正式公布 将于今年十二月一日起施行
历经最高立法机关三次审议,十三届全国人大常委会第十一次会议6月29日表决通过了《中华人民共和国疫苗管理法》。《疫苗管理法》共十一章100条,对疫苗的研制、生产、流通、预防接种等各环节均作出了明确规定,该法自2019年12月1日起施行。这是我国对疫苗管理进行的专门立法,将对疫苗实行最严格的管理制度,坚持安全第一、风险管理、全程管控、科学监管、社会共治。
专家指出,这部疫苗管理的专门法律,回应了人民群众的期待,解决疫苗管理中存在的突出问题,在制度设计中充分体现了药品领域“四个最严”的要求。
作为管理法,法律的“牙齿”很重要。从2018年12月底全国人大常委会第一次审议到近日的第三次审议,有关疫苗违法犯罪行为的法律责任一直在“加码”。《疫苗管理法》明确,疫苗犯罪行为依法从重追究刑事责任;对违法生产销售假劣疫苗,违反生产、储存、运输相关质量管理规范要求等情形的,设置了比一般药品更高的处罚;落实“处罚到人”要求,依法实行罚款、行政拘留、从业禁止直至终身禁业等。
《疫苗管理法》还为疫苗管理的全链条、各环节、各主体都设定了严格的责任。有些是制度上的创新,比如国家将实行疫苗全程电子追溯制度、预防接种异常反应补偿制度和疫苗责任强制保险制度等。
有些是对原有措施的“升级”。在生产环节,《疫苗管理法》提出,国家对疫苗生产实行严格准入制度。从事疫苗生产活动,要在《药品管理法》规定的从事药品生产条件之外,满足更加严格的条件。
在流通环节,《疫苗管理法》明确,疫苗储存、运输的全过程应当处于规定的温度环境,冷链储存、运输应当符合要求,并定时监测、记录温度。对于违反上述要求的单位和个人,将给予没收所得、罚款等惩罚。
在预防接种环节,《疫苗管理法》对接种单位的设置、人员资质及冷链作出严格规定,并要求医疗卫生人员在接种前、接种时、接种后严格按照要求提供预防接种服务,比如接种时要“三查七对”,接种后发现不良反应要及时救治等。
在监督管理环节,《疫苗管理法》提出,国家建设中央和省级两级职业化、专业化药品检查员队伍;疫苗管理部门要建立质量、预防接种等信息共享机制;实行疫苗安全信息统一公布制度等。
原文:http://www.npc.gov.cn/npc/xinwen/2019-06/29/content_2090368.htm
来源:中国人大网
监管动态
CDE发布中医药产业3项技术指导原则
6月28日,CDE发布了震动中医药产业的3项技术指导原则,分别对中药材、中药饮片、中药制剂三个方面制定了质控指标和检测方法的依据。
为深化药品审评审批制度改革,进一步鼓励中药创新研发,加快建立符合中药特点的技术评价标准体系,药品审评中心自2018年起启动了12个中药药学研究技术指导原则的制修订工作。同时,为配合国家药品监督管理局《中药饮片质量集中整治工作方案》(国药监〔2018〕28号)的实施,引导加强和规范中药材、饮片、制剂的质量管理,药品审评中心组织专家撰写了《中药材质量控制研究技术指导原则(征求意见稿)》、《中药新药质量标准研究技术指导原则(征求意见稿)》、《中药原料前处理技术指导原则(征求意见稿)》(详见附件1-3)。现上网公开征求意见,欢迎社会各界提出宝贵意见和建议。
原文:http://www.cde.org.cn/news.do?method=viewInfoCommon&id=314885
来源:CDE网站
国家药监局:医疗器械唯一标识系统试点工作方案
为加强医疗器械全生命周期管理,提升医疗器械监管和卫生管理效能,进一步保障公众用械安全,国家药品监督管理局会同国家卫生健康委员会开展医疗器械唯一标识系统试点工作。
原文:http://www.nmpa.gov.cn/WS04/CL2197/338683.html
来源:国家药监局
综合分析
2019年6月CDE药品审评情况报告
摘要
· 6月CDE共承办药品注册申请628件
· 恒瑞医药提交马来酸吡咯替尼新适应症临床申请
· 正大天晴首家提交甲磺酸仑伐替尼胶囊仿制申请
· 9个品种通过仿制药一致性评价
· 69个受理号获临床试验默示许可
总体承办情况:药品注册申请628个
据MED中国药品审评数据库2.0统计,2019年6月CDE共承办药品注册申请628个。除新增新药申请比上月有所上升外,仿制申请、进口申请以及补充申请(一致性评价)申请相比上月均有所回落。
详细信息请见:http://www.menet.com.cn/info/201907/201907040932403240_138965.shtml
来源:米内网
一致性评价最新进展:受理号达1197个,通过216个,视同通过77个
截止2019年6月26日,CDE承办的一致性评价受理号已达1197个,共计350个品种,涉及368家药企;其中,289目录品种受理号590个,共计129个品种。注射剂受理号297个,共计97个品种,涉及72家药企。
详细信息请见:https://news.yaozh.com/archive/26574.html
来源:药智网
盘点:2019年上半年,中国1类新药IND数量57个
2019上半年新药注册1类IND总体情况
2019年上半年,国内1类新药(国产化药)注册申报IND品种约57个,创新品种开发形势整体处于增长态势,其中上半年注册申报数量最多的月份为1月,共16个品种进入IND阶段。
详细信息请见:https://news.yaozh.com/archive/26607.html
来源:药智网
国外信息
盘点:2019年1-6月,FDA批准的8个NME!
FDA对于药品的评价,一直都是全球药学相关研究的关注热点,而NME的获批上市,更是重中之重。2019年1-6月,FDA共批准8个NME上市,其中包括针对适应症的首个药物、首个靶点抑制剂。
2019年1-6月FDA批准的8个NME
FDA对于药品NDA的提交分类不同于我国,其共有十余种,详情见表1。其中,Type1-5关注度较高,尤其是Type1-NME(New molecular entity),关注度最高!
详细信息请见:https://news.pharmacodia.com/news/html/info/info-detail.html?id=39946
来源:药渡
盘点2019年上半年获批新药中的“首例”
根据FDA药物评估和研究中心(CDER)的数据统计,2019年上半年,FDA总计批准了13款创新药。这一统计并不包括基因和细胞疗法。而FDA生物制剂评估和研究中心(CBER)的数据表明,2019年上半年,FDA还批准了一款基因疗法和一款疫苗。今年获批的新药中有多个“首例”。
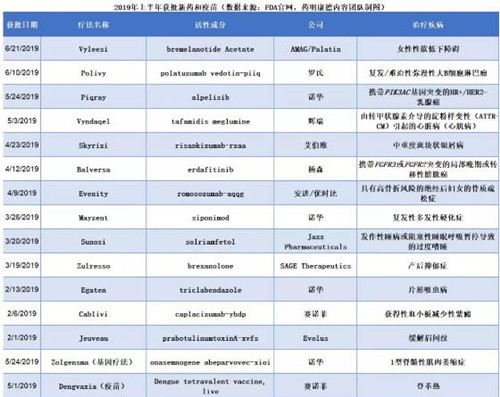
▲2019年上半年获批新药和疫苗(数据来源:FDA官网,药明康德内容团队制图)
首例通过实时肿瘤学审评(RTOR)获批的新分子实体
2018年FDA创新药获批数目能够创纪录,不但是新药研发企业努力的结果,也是FDA锐意改革,加快新药审评速度的结果。而RTOR,是FDA属下肿瘤学卓越中心推出的,帮助加快肿瘤学药物审评速度的一个重要试点项目。这一项目允许FDA在正式申请递交之前获得关键性数据,让审评团队能够更早开始审评过程并且与申请人进行沟通。
这一试点项目已经用于批准多款抗癌疗法扩展适应症。而今年5月,它第一次被用于批准诺华公司开发的Piqray(alpelisib)。使用RTOR和其它FDA推出的新举措,Piqray的获批时间比预计的PDUFA日提前了接近3个月!
将新药以最快的速度交到患者的手中,离不开监管部门的支持和协助,我们期待FDA继续进行监管流程的现代化,为造福更多患者助力。
首例治疗乳腺癌的PI3K抑制剂和首例治疗膀胱癌的个体化疗法
在癌症治疗领域,精准疗法正在逐渐成为新药研发的一个重要方向。去年,靶向NTRK基因融合的Vitrakvi是精准疗法的范例之一。而今年上半年获批的三款抗肿瘤疗法中,有两款创新疗法靶向携带特定基因突变的癌症患者。上面提到的Piqray是第一款针对携带PIK3CA基因突变的HER2阴性乳腺癌患者的PI3K抑制剂。而4月获批的Balversa(erdafitinib)是针对携带致敏性FGFR3或FGFR2基因突变的膀胱癌患者的FGFR激酶抑制剂。这两款创新疗法都需要患者接受FDA批准的伴随检测,确认患者携带致敏性基因突变。
首例治疗脊髓性肌肉萎缩症的基因疗法
基因疗法的复兴和蓬勃发展,是近两年来创新疗法开发的主题之一。今年9月17日,是Jesse Gelsinger先生去世20周年,这位年轻的罕见遗传病患者在接受基因疗法治疗的临床试验中,因为对病毒载体的免疫反应而去世。他的去世让基因疗法领域的研发停滞了近10年。曾经,人们以为基因疗法无法从这一悲剧的阴影中走出来。但是在科研人员的不懈努力之下,基因疗法领域不但获得重生,而且近年来获得大型药企广泛关注。
治疗脊髓性肌肉萎缩症(SMA)的Zolgensma为基因疗法的复兴提供了一个最好的注释。这款基因疗法的研发过程也受到了Gelsinger先生去世的影响,然而研发人员的坚持,与患者勇敢的支持,让这款挽救SMA患者生命,并且为他们的未来带来无限可能的创新疗法终于获得FDA的批准上市。首例治疗产后抑郁症的创新疗法。
在美国,每9位妇女中就有一位受到产后抑郁症的困扰。这些患者在今年3月终于迎来的第一款专门针对产后抑郁症的创新疗法。这一创新药物研发的起点源于上世纪80年代在美国国家心理健康研究所(NIMH)的基础科学研究。研究人员发现,人体中的黄体酮(progesterone)和去氧皮质酮的代谢产物能够与大脑中的抑制性神经递质GABA的受体结合。这些代谢产物能够增强GABA的抑制功能,从而影响神经细胞的兴奋性。
这一发现催生的一系列基础研究发现,这些代谢物的水平随着月经周期起伏。其中名为别孕烯醇酮的代谢物水平在孕期升高,在分娩后快速下降。而这是导致某些女性在分娩后出现抑郁和焦虑的原因之一。
Sage Therapeutics公司根据这些发现,开发出一种别孕烯醇酮配方,能够恢复产后妇女体内的激素水平。这款拥有创新作用机制的新药最终成为今年3月获批的Zulresso,为这一产品从学术研究转为创新疗法的征程画上了圆满的句号。
治疗抑郁症的新药研发一直是一个重大挑战,以前的抗抑郁药物作用的信号通路多为血清素信号通路。而今年除了Zulresso获得FDA批准以外,杨森(Janssen)公司的Spravato(esketamine)也获得FDA批准治疗严重抑郁症(由于ketamine曾经获批治疗其它适应症,Spravato不被列为新分子实体)。这款疗法同样具有与以往抗抑郁药物不同的作用机制,靶向NMDA信号通路。我们期待具有创新作用机制的抗抑郁药物能够不断涌现,开创治疗抑郁患者的新时代。
结语
虽然上半年获批新药的数目与创纪录的2018年(上半年20款新药获批)相比还有些差距,但是我们看到具有创新机制的新药仍然不断涌现,FDA对药物审评过程的改革和对新药研发的支持仍然不变。在今年的下半年中,我们预计还将看到多款具有创新机制的新药获得FDA批准,其中包括第二款“不限癌种”,靶向NTRK基因融合的精准疗法Rozlytrek(entrectinib)。这款新药已经在日本首先获批上市。
除此之外,新基公司的创新贫血疗法luspatercept,SAREPTA Therapeutics治疗杜兴氏肌营养不良症(DMD)的外显子跳跃疗法golodirsen,Aimmune Therapeutics公司治疗花生过敏的AR101都代表着治疗各自适应症方面的创新机制。
来源:药明康德
盘点7月将面临美国FDA重要监管决定的5个新药
到目前为止,今年1月至6月,美国FDA仅批准了12个新分子实体,而去年同期有多达20个。知名财经网站RTTNews近日发文,7月将有5款药物在美国监管方面迎来重要审查决定。以下是每个药物的相关情况:
1、selinexor
7月6日,FDA将对Karyopharm Therapeutics公司靶向抗癌药selinexor的新药申请(NDA)作出审查决定。该申请寻求FDA加速批准selinexor与地塞米松联合用药方案,用于既往已接受至少3种疗法且其疾病对至少一种蛋白酶体抑制剂(PI)、一种免疫抑制剂(IMiD)、一种抗CD38单克隆抗体难治的复发难治性多发性骨髓瘤(RRMM)患者。来自IIb期临床研究STORM的数据显示,selinexor与地塞米松联合用药方案的总缓解率(ORR)为26.2%,中位总生存期为8.6个月。
值得注意的是,今年2月底,FDA肿瘤药物咨询委员会(ODAC)召开会议对selinexor进行了讨论和表决。经过对NDA数据仔细审查后,ODAC以8票否定、5票赞成的投票结果,不支持加速批准selinexor。ODAC同时建议FDA应在关键性III期临床研究BOSTON的数据出炉之后再做审批决定。目前,该研究正在进行中,预计2019年底或2020年初获得顶线数据。
ODAC的建议对FDA并不具有约束力,但FDA在做出最终审查时通常都会考虑其意见。之前,FDA已授予selinexor孤儿药资格和快速通道资格。
selinexor是一种首创、口服、选择性核输出抑制剂(SINE)化合物,通过结合并抑制核输出蛋白XPO1发挥作用,导致肿瘤抑制蛋白在细胞核内积累,这将重新启动并放大它们的肿瘤抑制功能,导致癌细胞选择性凋亡,同时不会对正常细胞造成显著影响。目前,Karyopharm也正在多个中后期临床研究中评估selinexor治疗一系列血液系统恶性肿瘤和实体瘤的潜力。
2、relebactam
7月16日,FDA将对默沙东抗生素relebactam的新药申请做出审查决定,该申请寻求批准relebactam与亚胺培南/西司他丁(imipenem/cilastatin)固定剂量组合,用于成人患者治疗由某些易感革兰氏阴性菌导致的感染。在美国,亚胺培南/西司他丁以品牌名Primaxin销售,这是一种常用于治疗多种细菌性感染的有效抗生素产品。
relebactam是一种新型β-内酰胺酶抑制剂,属于二氮杂双环辛烷抑制剂,具有广谱抗β内酰胺酶活性,包括 A 类(超广谱β内酰胺酶和 KPC)和C类(AmpC 酶)。针对亚胺培南耐药的革兰氏阴性菌株,联合应用relebactam时,菌株会对亚胺培南变得更加敏感。此前,FDA已授予relebactam合格传染病产品(QIDP)资格和快速通道资格。
在关键性III期研究RESTORE-IMI 2中,与Colistin(多粘菌素E)+亚胺培南/西司他丁组合方案相比,relebactam+亚胺培南/西司他丁组合方案可有效治疗亚胺培南非敏感性细菌感染(主要终点),此外在治疗中还具有较低的肾毒性(次要终点)。
3、Otezla
7月21日,FDA将对新基重磅药物Otezla(apremilast)治疗白塞氏病(Behcet's disease,BD)的扩大适应症作出审查决定。BD是一种罕见的自身免疫性疾病,是以小血管炎为病理基础的多系统受累全身性疾病,以口腔、生殖器、皮肤及眼部受累最为常见。临床典型表现为眼-口-生殖器三联症,即反复发作性口腔溃疡、眼色素膜炎及生殖器溃疡。
Otezla的活性药物成分为apremilast,这是一种口服小分子磷酸二酯酶(PDE4)抑制剂,在细胞内调控促炎症和抗炎介质的网络。PDE4是一种环磷酸腺苷(cAMP)特异性PDE,是炎性细胞中主要的PDE。PDE4抑制可提升细胞内cAMP水平,通过调控TNF-α、IL-23和其他炎性细胞因子的表达相应下调炎性反应。cAMP升高也会增加抗炎细胞因子,例如IL-10。
Otezla已被批准用于治疗银屑病和银屑病关节炎,2018年销售额达16.1亿美元,较前一年增长26%。目前,百时美施贵宝正在收购新基,不过这笔高达740亿美元的并购案进展颇不顺利。
就在近日,百时美宣布已与美国联邦贸易委员会(FTC)达成一项裁决意见,计划出售Otezla。由于此次裁决,这笔原计划今年三季度完成的并购交易,其完成时间可能会推迟至2019年底或2020年初。
有分析师指出,Otezla在银屑病领域与多达5-7个药物展开竞争,但并未被视为市场的“主导力量”。此次FTC的裁决,表明该机构在医药并购方面正在加大对竞争的监管力度。
4、Ofev
7月25日,FDA专家小组将对勃林格殷格翰Ofev(nintedanib)治疗系统性硬化症相关间质性肺病(SSc-ILD)的补充新药申请做出审查建议。
系统性硬化症(SSc)也被称为硬皮症,是一种罕见的、不可治愈的、影响全身结缔组织的自身免疫性疾病,可引起皮肤、心、肺、消化道、肾等主要器官瘢痕(纤维化),并可能有危及生命的并发症。当SSc影响到肺部时可导致间质性肺病(ILD),即SSc-ILD。据估计,约80%的SSc患者病情会发展至ILD,这是导致SSc患者死亡的关键因素,约占死亡病例的三分之一。目前,尚无治疗SSc-ILD的药物。
Ofev的活性药物成分为nintedanib,这是一种口服小分子酪氨酸激酶抑制剂,能够抑制多种受体酪氨酸激酶(RTK)和非受体酪氨酸激酶(nRTK)。通过竞争性抑制成纤维细胞生长因子受体(FGFR 1-3)、血管内皮生长因子受体(VEGFR 1-3)、血小板源性生长因子受体(PDGFR α和β)等受体酪氨酸激酶,该药可阻断对成纤维细胞的增殖、迁徙和转换起关键作用的信号传导,从而抑制肺纤维化病变。所有这3类受体在血管生成和肿瘤生长过程中也发挥着重要作用。
截止目前,nintedanib已获全球70多个国家批准治疗特发性肺纤维化(IPF),以品牌名Ofev销售。此外,该药也已获部分国家批准治疗非小细胞肺癌(NSCLC),以品牌名Vargatef销售。在2018年,该药的销售额增长超过30%,达到11亿欧元。SSc-ILD将成为该药的第3个适应症,有望为本已强劲的销售增长提供更多动力。
5、lumateperone
7月31日,FDA专家小组将对IntraCellular Therapies公司lumateperone治疗成人精神分裂症的新药申请做出审查建议。该药是一种每日口服一次的药物,具有一种新颖的作用机制。之前,FDA已授予该药快速通道资格。
lumateperone是一种首创的(first-in-class)小分子药物,可选择性且同时调节5-羟色胺、多巴胺及谷氨酸这3种涉及严重疾病的神经递质通路。
与现有的精神分裂症药物不同,lumateperone是一种多巴胺受体磷酸蛋白调节剂(DPPM),在D2受体上充当突触前部分激动剂和突触后拮抗剂,这种机制,连同与5-HT2A受体、5-羟色胺转运体及D1受体的潜在相互作用以及间接的谷氨酸调节作用,可能有助于lumateperone在横跨一系列精神症状方面的疗效,具有改善的心理社会功能和良好的耐受性。这种化合物有可能使罹患一系列神经精神障碍和神经退行性疾病的患者受益。
来源:新浪医药新闻
欧盟《生产商符合欧盟防伪法令的GMP检查备忘录》(中英文)
近日,欧盟委员会发布了《生产商符合欧盟防伪法令(EU)2016/161的GMP检查备忘录》,该GMP检查备忘录包括用以确认生产商符合欧盟防伪法令(EU)2016/161需要审查的问题,以确保在检查之前符合该法律。该法规概述了医药产品的安全特性,特别是唯一标识符(UI)的特征和技术规格,安全特征验证的方式以及包含安全特征信息的储存库系统的管理。
欧盟委员会已提醒制药公司,他们将检查工厂生产的所有要求具备安全功能产品是否符合法令要求。备忘录包括有关数据流的要求,特别是有关如何生成序列号的要求,打印UI的位置以及UI的上传。备忘录中还详细介绍了与包装线,UI组成,UI注销,人类可读格式,二维码打印质量,警报管理以及篡改或疑似伪造时所需的操作相关的要求。
中英文全文下载:欧盟法令(EU)2016 161 安全特性的生产商符合性 GMP检查备忘录(中英文).pdf
来源:GMP办公室
欧盟发布《序列化追溯系统GMP检查指南》
欧盟防伪法令要求自2019年2月9日起具备安全特性(序列化追溯+防篡改装置)
目前来看,已有不少公司因安全特性(序列化追溯+防篡改装置)落下严重缺陷!
近日,欧盟委员会发布了一份《欧盟法令(EU)2016/161安全特性的GMP符合性检查备忘录》,列出了GMP检查过程中特别关注的9类问题:
﹒与OBP的连接
﹒数据流
﹒序列码(SN)的生成
﹒数据上传
﹒防篡改装置(ATD)的应用
﹒包装线
﹒唯一标识符的状态更改
﹒二维条码打印的质量
﹒报警管理
例如,关于序列码生成、数据上传和包装线方面,在GMP检查过程中将确认以下方面:
Generation of Serial Numbers (SNs)
序列码的生成
Where/by whom are the SNs generated? Is there a Contract in place?
在哪里/谁生成了序列码?是否有合同?
Is it generated by a deterministic or a non- deterministic randomisation algorithm, in a way that the probability that the serial number can be guessed shall be negligible and in any case lower than one in ten thousand?
它是由确定性或非确定性随机化算法生成的,可猜到序列号的概率可以忽略不计,并且在任何情况下都低于万分之一?
Is the combination of the PC+SN unique until EXP+1Y or REL+5Y, whichever is the longer period?
PC+SN 的组合在有效期后一年( EXP+1Y) 或五年( REL=5Y)(以较长的周期为准) 之前是否唯一?
Is serialisation data received from other parties, e.g. CMO’s? If yes, how (e.g. connection with the CMO’s system)?
是否从其他方(例如 CMO)接收序列化数据?如果是,如何接收?(例如:如何与 CMO 的系统连接)?
Has the security of the connection been evaluated?
连接的安全性是否已评估?
Who manages/controls the Product Master Data in the hub (e.g. creation of a new product, changes to an existing product)?
谁管理/控制中心中的主数据(例如,创建新产品、对现有产品的更改)?
How is it ensured that only Product Master Data from legitimate marketed packs is uploaded?
如何确保仅上传合法销售包中的产品主数据?
(i.e. once a company passes EMVO’s legitimacy check and gets access, how is that company prevented from creating non-existing products in the system and upload of SN’s for t his fake product, to enable distribution of falsified product)
(例如,一旦公司通过 EMVO 的合法性检查并获得访问权限,该公司如何阻止在系统中创建不存在的产品并上传假冒产品SN
Uploading of information in the repositories system
在存储库系统中上传信息
At what point in the batch release process is the data uploaded?
在批放行过程中的什么节点上传数据?
Is the data sent to the serialisation partner’s server first and held for a period or stored temporarily in the manufacturer’s/MAH’s cloud, prior to upload to the hub?
在上传到数据中心之前,数据是否首先发送到序列化合作伙伴的服务器,并暂存一段时间或暂存在生产企业/MAH的云服务器中?
How is the upload to the hub actually triggered?
如何触发上传到数据中心?
How is it ensured that only the data for ‘good’ packs (suitable for release) is uploaded to the hub?
如何确保只有"好的"包装(可放行)的数据上传到数据中心?
Is the system designed in a way that no upload of data goes undetected/that any upload of data requires approval (of the QP?) before actually sending it to the hub?
系统的设计方式是否是:数据不会未经检测即上传/即任何数据上传都需要(经过QP)批准?
What happens to the UIs which were generated but not used and UIs on packs ejected from the line at the eject stations during packaging?
如何处理已生成但未使用的 UIs 和在包装过程被从生产线剔除的包装上的UIs?
Is a verification of successful upload and distribution required to be obtained?
是否需要验证成功上传和分发?
Is it verified whether the quantity of serial numbers successfully received by the NMVS, corresponds to the quantity of serial numbers that was initially intended to be uploaded (reconciliation of the number of SN’s)?
是否验证 NMVS 成功接收的序列码的数量是否与最初上传的序列码数量一致(SN 编码的物料平衡)?
Who receives this and what action is required in the event of a failed upload?
谁收到此内容,上传失败时需要执行采取什么措施?
Does the (successful) upload occur before or after batch certification by the QP?
(成功)上传是在 QP 的批放行之前还是之后?
Does the (successful) upload happen before or after release to the market or for export?
(成功)上传是在放行上市/放行出口之前还是之后?
Are there procedures which describe these processes?
是否有描述这些流程的文件?
(Note: The information laid down in Article 33(2) of Commission Delegated Regulation (EU) 2016/161 needs to be present in the system at the time the batch is released for sale and distribution)
(注:2016/161年《委员会委托条例》(EU)第33(2)条中规定,在放行销售和分销时,信息需要存在于系统中)
Packaging Lines
包装线
Was serialisation for EU implementedat the site under change control?
欧盟序列化在变更控制下实施吗?
Did this change controlprocess include identification of QMS documentation which required update toincorporate safety features?(e.g. procedures for recall,quality defects, batch disposition, shipment, distribution etc.; batch records,job descriptions for key personnel/QP, technical/quality agreements)
此变更控制流程是否包括需要更新以纳入安全功能的 QMS 文件的标识?(例如召回程序、质量缺陷、批次处置、装运、分销等;批记录、关键人员/QP的岗位说明、技术/质量协议)
Are both the 2D barcode andthe ATD applied?
二维码和防篡改装置都应用了吗?
In the case of the ATD, getthe company to demonstrate that if removed or broken, this is evident visuallyfrom the pack.
防篡改装置让公司可以从包装上明显的看出开封或损坏。
Is stability data available?(Tamper evident nature shouldbe proven throughout the shelf life of the pack)
稳定性数据是否可用?(在包装的效期内应开盒可留痕)
Which packaging lines havecapability for serialisation?
哪些包装生产线具有序列化功能?
Was new equipment installedand was it qualified? (e.g. printers, cameras, reject stations etc.).
新设备安装后是否经设备验证?(例如打印机、摄像头、剔除等)。
Review change control,qualification documentation etc.
审核变更控制、设备确认文件等
Is the UI printed on the packsonline or are labels/stickers applied to packs separately?
UI 是在线打印在包装上,还是将标签/标签分开贴在包装上?
Is there 100% verification ofthe readability of the 2D barcode? How is this done (e.g. on-line camera)?
二维条码的可读性是否100%确认?这是如何做到的(例如在线摄像机)?
Is there an on-line sensor todetect the presence of ATDs and is it challenged?
是否有在线探头来检测是否存在 ATD,是否有挑战?
Is aggregation implemented?Explain how (e.g. UI’s in 1data-file. What happens with the data-file. How is this protected/transferredin a secure way)?
是否实现打包整合?说明如何打包(例如 UI 位于 1 个数据文件中)。数据文件会发生什么情况。如何以安全的方式保护/传输?
原文下载:欧盟产品序列化追溯系统GMP检查指南_en.pdf
来源:GMP办公室
欧盟/美国GMP互认:德国最终被确认为第27个国家!
2019年6月26日,德国被纳入欧盟(EU)和美国食品和药物管理局(FDA)互认协议。因此,FDA确认了德国有能力进行相当于美国的GMP检查。该协议相互承认对在不同领土进行的人用药生产基地的检查,这意味着FDA将依靠欧盟27个会员国,其检查结果等同FDA检查。

目前通过欧美互认协议的国家包括:德国、卢森堡、荷兰、保加利亚、塞浦路斯、波兰、斯洛文尼亚、比利时、丹麦、爱沙尼亚、芬兰、拉脱维亚、葡萄牙、、爱尔兰、立陶宛;、捷克共和国、希腊、匈牙利、罗马尼亚;、奥地利、克罗地亚、法国、意大利、马耳他、西班牙、瑞典和英国。
目前欧盟成员仅剩斯洛伐克仍未进入此行列。按照美国和欧盟之间的MRA目标:到2019年7月15日,将完成对所有28个欧盟成员国的确认。
来源:GMP办公室