本 期 要 目
[监管动态]
药品检查员队伍向职业化专业化迈进——国务院办公厅印发《关于建立职业化专业化药品检查员队伍的意见》
国家药监局发布关于进一步完善药品关联审评审批和监管工作有关事宜的公告
国家药监局发布修订西达本胺片说明书的公告
[综合分析]
2019年国内临床试验年中报告,一致性评价BE试验占19%
仿制药一致性评价最新进展:受理号达1289个,通过308个
2019上半年24个创新器械产品进入CMDE“绿色通道” 国产超七成
[国外信息]
2009-2018年六大监管机构的新药批准情况
FDA发布《辅料数据库使用指南》草案
2018年FDA数据完整性相关警告信分析
日本医疗器械监管概览
监管动态
药品检查员队伍向职业化专业化迈进——国务院办公厅印发《关于建立职业化专业化药品检查员队伍的意见》
近日,国务院办公厅印发《关于建立职业化专业化药品检查员队伍的意见》(以下简称《意见》)。
《意见》指出,要以习近平新时代中国特色社会主义思想为指导,按照党中央、国务院关于加强药品安全监管的决策部署,遵循科学监管规律,深化药品监管体制机制改革,坚持源头严防、过程严管、风险严控,强化药品安全监督检查,切实保障人民群众身体健康和用药用械安全。
《意见》提出,职业化专业化药品(含医疗器械、化妆品)检查员是指经药品监管部门认定,依法对管理相对人从事药品研制、生产等场所、活动进行合规确认和风险研判的人员,是加强药品监管、保障药品安全的重要支撑力量。要坚持职业化方向和专业性、技术性要求,到2020年底,国务院药品监管部门和省级药品监管部门基本完成职业化专业化药品检查员队伍制度体系建设。在此基础上,再用三到五年时间,构建起基本满足药品监管要求的职业化专业化药品检查员队伍体系,进一步完善以专职检查员为主体、兼职检查员为补充,政治过硬、素质优良、业务精湛、廉洁高效的检查员队伍。
《意见》提出了五方面政策措施。一是完善药品检查体制机制。构建国家、省两级职业化专业化药品检查员队伍,强化检查机构建设,明确检查事权划分,落实检查要求,完善检查工作协调机制。二是落实检查员配置。合理确定队伍规模,规范检查员编制管理,创新检查员管理机制,多渠道充实检查员队伍。三是加强检查员队伍管理。职业化专业化药品检查员实行分级分类管理,确立严格的岗位准入和任职条件,建立科学合理的考核评价与职级升降机制。四是不断提升检查员能力素质。强化检查员业务培训,鼓励检查员提升能力水平,创新高素质检查员培养模式。五是建立激励约束机制。拓宽检查员职业发展空间,完善检查员参加相应职称评审的政策,建立检查员薪酬待遇保障机制,强化纪律约束和监督。
意见原文:http://www.gov.cn/zhengce/content/2019-07/18/content_5411172.htm
来源:中国食品药品网
国家药监局发布关于进一步完善药品关联审评审批和监管工作有关事宜的公告
为落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),原食品药品监管总局发布了《关于调整原料药、药用辅料和药包材审评审批事项的公告》(2017年第146号),现就进一步明确原料药、药用辅料、直接接触药品的包装材料和容器(以下简称原辅包)与药品制剂关联审评审批和监管有关事宜公告如下:
一、总体要求
(一)原辅包的使用必须符合药用要求,主要是指原辅包的质量、安全及功能应该满足药品制剂的需要。原辅包与药品制剂关联审评审批由原辅包登记人在登记平台上登记,药品制剂注册申请人提交注册申请时与平台登记资料进行关联;因特殊原因无法在平台登记的原辅包,也可在药品制剂注册申请时,由药品制剂注册申请人一并提供原辅包研究资料。
(二)原辅包登记人负责维护登记平台的登记信息,并对登记资料的真实性和完整性负责。境内原辅包供应商作为原辅包登记人应当对所持有的产品自行登记。境外原辅包供应商可由常驻中国代表机构或委托中国代理机构进行登记,登记资料应当为中文,境外原辅包供应商和代理机构共同对登记资料的真实性和完整性负责。
(三)药品制剂注册申请人申报药品注册申请时,需提供原辅包登记号和原辅包登记人的使用授权书。
(四)药品制剂注册申请人或药品上市许可持有人对药品质量承担主体责任,根据药品注册管理和上市后生产管理的有关要求,对原辅包供应商质量管理体系进行审计,保证符合药用要求。
(五)监管部门对原辅包登记人提交的技术资料负有保/密责任,对登记平台的技术信息保/密,登记平台只公开登记品种的登记状态标识(A或I)、登记号、品种名称、企业名称(代理机构名称)、企业生产地址、原药品批准文号(如有),原批准证明文件有效期(如有),产品来源、规格、更新日期和其他必要的信息。
二、产品登记管理
(六)原辅包登记人按照登记资料技术要求在平台登记,获得登记号。其中,原料药在登记前应取得相应生产范围的《药品生产许可证》,并按照原食品药品监管总局《关于发布化学药品新注册分类申报资料要求(试行)的通告》(2016年第80号)要求进行登记;药用辅料和药包材登记按照本公告附件1、附件2的资料要求进行登记。登记资料技术要求根据产业发展和科学技术进步不断完善,由国家药品监督管理局药品审评中心(以下简称药审中心)适时更新公布。
(七)药品制剂注册申请与已登记原辅包进行关联,药品制剂获得批准时,即表明其关联的原辅包通过了技术审评,登记平台标识为“A”;未通过技术审评或尚未与制剂注册进行关联的标识为“I”。
(八)除国家公布禁止使用、淘汰或者注销的原辅包外,符合以下情形的原辅包由药审中心将相关信息转入登记平台并给予登记号,登记状态标识为“A”:
1.批准证明文件有效期届满日不早于2017年11月27日的原料药;
2.已受理并完成审评审批的原料药,含省局按照国食药监注〔2013〕38号文审评的原料药技术转让申请;
3.已受理并完成审评的药用辅料和药包材;
4.曾获得批准证明文件的药用辅料;
5.批准证明文件有效期届满日不早于2016年8月10日的药包材。
转入登记平台的原辅包登记人应按照本公告登记资料要求在登记平台补充提交研究资料,完善登记信息,同时提交资料一致性承诺书(承诺登记平台提交的技术资料与注册批准技术资料一致)。
(九)仿制或进口境内已上市药品制剂所用的原料药,原料药登记人登记后,可进行单独审评审批,通过审评审批的登记状态标识为“A”,未通过审评审批的标识为“I”。审评审批时限和要求按照现行《药品注册管理办法》等有关规定执行。
(十)已在食品、药品中长期使用且安全性得到认可的药用辅料可不进行登记(名单详见附件3),由药品制剂注册申请人在制剂申报资料中列明产品清单和基本信息。但药审中心在药品制剂注册申请的审评过程中认为有必要的,可要求药品制剂注册申请人补充提供相应技术资料。该类药用辅料品种名单由药审中心适时更新公布。
(十一)药用辅料、药包材已取消行政许可,平台登记不收取费用。原料药仍为行政许可,平台登记技术审评相关要求按现行规定和标准执行。
三、原辅包登记信息的使用和管理
(十二)药品制剂注册申请关联审评时,原辅包登记平台研究资料不能满足审评需要的,药审中心可以要求药品制剂注册申请人或原辅包登记人进行补充。补充资料的报送途径由药审中心在发补通知中明确。
(十三)原料药标识为“A”的,表明原料药已通过审评审批。原料药登记人可以在登记平台自行打印批准证明文件、质量标准和标签等,用于办理GMP检查、进口通关等。
未进行平台登记而与药品制剂注册申报资料一并提交研究资料的原料药,监管部门在药品制剂批准证明文件中标注原料药相关信息,可用于办理原料药GMP检查、进口通关等。
(十四)原料药生产企业申请GMP检查程序及要求按照现行法律法规有关规定执行,通过药品GMP检查后应在登记平台更新登记信息。
(十五)标识为“A”的原料药发生技术变更的,按照现行药品注册管理有关规定提交变更申请,经批准后实施。原料药的其他变更、药用辅料和药包材的变更应及时在登记平台更新信息,并在每年第一季度提交的上一年年度报告中汇总。
(十六)原辅包发生变更时原辅包登记人应主动开展研究,并及时通知相关药品制剂生产企业(药品上市许可持有人),并及时更新登记资料,并在年报中体现。
药品制剂生产企业(药品上市许可持有人)接到上述通知后应及时就相应变更对药品制剂质量的影响情况进行评估或研究,属于影响药品制剂质量的,应报补充申请。
(十七)已上市药品制剂变更原辅包及原辅包供应商的,应按照《已上市化学药品变更研究技术指导原则(一)》《已上市化学药品生产工艺变更研究技术指导原则》《已上市中药变更研究技术指导原则(一)》及生物制品上市后变更研究相关指导原则等要求开展研究,并按照现行药品注册管理有关规定执行。
(十八)境外原辅包供应商更换登记代理机构的,提交相关文件资料后予以变更。包括:变更原因说明、境外原辅包供应商委托书、公证文书及其中文译本、新代理机构营业执照复印件、境外原辅包供应商解除原代理机构委托关系的文书、公证文书及其中文译本。
四、监督管理
(十九)各省(区、市)药品监督管理局对登记状态标识为“A”的原料药,按照药品进行上市后管理,并开展药品GMP检查。
(二十)各省(区、市)药品监督管理局应加强对本行政区域内药品制剂生产企业(药品上市许可持有人)的监督检查,督促药品制剂生产企业(药品上市许可持有人)履行原料药、药用辅料和药包材的供应商审计责任。
药用辅料和药包材生产企业具有《药品生产许可证》的,继续按原管理要求管理,许可证到期后按本公告要求登记场地信息。
(二十一)各省(区、市)药品监督管理局根据登记信息对药用辅料和药包材供应商加强监督检查和延伸检查。发现药用辅料和药包材生产存在质量问题的,应依法依规及时查处,并要求药品制剂生产企业(药品上市许可持有人)不得使用相关产品,并对已上市产品开展评估和处置。延伸检查应由药品制剂生产企业(药品上市许可持有人)所在地省局组织开展。药用辅料和药包材供应商的日常检查由所在地省局组织开展联合检查。
药用辅料生产现场检查参照《药用辅料生产质量管理规范》(国药监安〔2006〕120号)开展检查,药包材生产现场检查参照《直接接触药品的包装材料和容器管理办法》(原国家食品药品监督管理局局令第13号)中所附《药包材生产现场考核通则》开展检查。各省(区、市)药品监督管理局可根据监管需要进一步完善相关技术规范和检查标准,促进辅料和药包材质量水平稳步提升。
国家药品监督管理局将根据各省监督检查开展情况和需要,适时修订相关检查标准。
五、其他
(二十二)在中华人民共和国境内研制、生产、进口和使用的原料药、药用辅料、药包材适用于本公告要求。
(二十三)本公告自2019年8月15日起实施。原发布的原辅包相关文件与本公告要求不一致的,以本公告为准。原食品药品监管总局发布的《关于发布药包材药用辅料申报资料要求(试行)的通告》(2016年第155号)同时废止。
原文:http://www.nmpa.gov.cn/WS04/CL2138/339042.html
来源:国家药监局
国家药监局发布修订西达本胺片说明书的公告
为进一步保障公众用药安全,国家药品监督管理局决定对西达本胺片说明书【不良反应】、【注意事项】等项进行修订。现将有关事项公告如下:
一、西达本胺片生产企业应依据《药品注册管理办法》等有关规定,按照西达本胺片说明书修订要求(见附件),提出修订说明书的补充申请,于2019年9月16日前报省级药品监管部门备案。
修订内容涉及药品标签的,应当一并进行修订;说明书及标签其他内容应当与原批准内容一致。在补充申请备案后6个月内对所有已出厂的药品说明书及标签予以更换。
西达本胺片生产企业应当对新增不良反应发生机制开展深入研究,采取有效措施做好使用和安全性问题的宣传培训,涉及用药安全的内容变更要立即以适当方式通知药品经营和使用单位,指导医师、药师合理用药。
二、临床医师、药师应当仔细阅读西达本胺片说明书的修订内容,在选择用药时,应当根据新修订说明书进行充分的效益/风险分析。
三、患者应严格遵医嘱用药,用药前应当仔细阅读说明书。
来源:国家药监局
综合分析
2019年国内临床试验年中报告,一致性评价BE试验占19%
1.药物类别
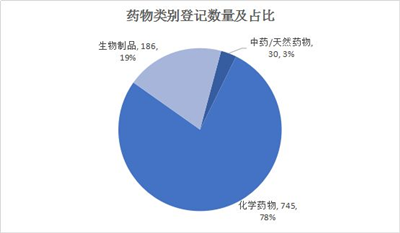
2019年上半年,CDE药物临床试验登记平台总共公示961个临床试验。其中,化学药物745个,生物制品186个,中药/天然药物30个。
2.国内试验和国际多中心试验
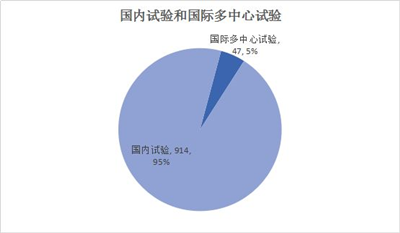
目前药物临床试验平台中的临床试验多以国内试验为主,2019年上半年总计47个国际多中心试验,大多都是3期试验,主要研究安全性和有效性。从药品来看,基本均为新药临床试验。值得一提的是,许多国内企业尤其是创新药企业开始重视国际多中心试验。如:青岛百洋制药有限公司、上海复宏汉霖生物技术股份有限公司、上海海和药物研究开发有限公司、益方生物科技(上海)有限公司、苏州爱美津制药有限公司、药华医药股份有限公司、百济神州(北京)生物科技有限公司、上海恒瑞医药有限公司、上海岸迈生物科技有限公司、百奥泰生物制药股份有限公司。
3.按月度登记(获得CTR号)的临床试验总数
临床试验登记数量来看,2019年上半年同比增加较多,其中4月更是达到了208个临床试验,与2018年5月的最高值207持平。

4.试验分类和分期
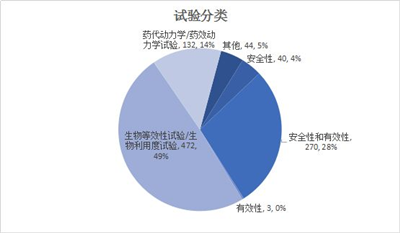
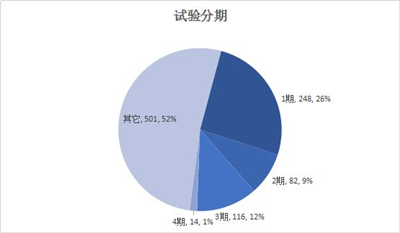
这里对试验分类和分期就不做太多分析,主要是因为一致性评价的开展,和常规临床试验登记规律有一定不同。
试验分类
试验分期
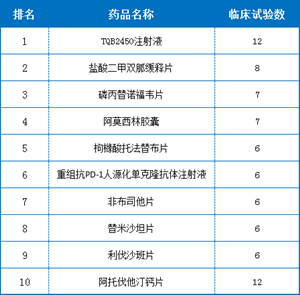
5.2019上半年临床试验最受关注的药品TOP10
对药品的临床试验登记情况统计发现,2019上半年最受关注的药品为TQB2450注射液,其次是盐酸二甲双胍缓释片。TQB2450注射液是正大天晴集团研发的创新型抗PD-L1单克隆抗体药物,由该公司附属公司南京顺欣制药有限公司申请,注册分类为治疗用生物制品。TQB2450注射液拥有结构、配方及工艺等多项自主知识财产权。
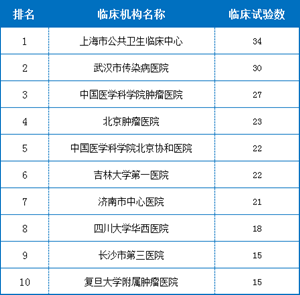
6.最忙碌的临床试验机构
简单看一下2019上半年最忙碌的临床试验机构,开展试验最多的是上海市公共卫生临床中心。与去年不同点在于,去年一致性评价的品种更多,所以很多医院接的临床试验出现了大幅度上升。
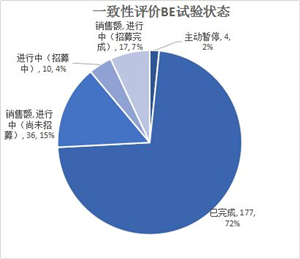
7.一致性评价BE试验
2019年上半年,仿制药一致性评价已登记的BE试验185个,比2018年同期下降24.18%。登记的BE试验主要格局如下:BE试验已经有177个完成了,同时还有15%尚未招募。
BE试验登记数量最多的药品分别是阿莫西林胶囊(9)、盐酸二甲双胍缓释片(5)和替米沙坦片(5)。
综上所述:纵观2019上半年已公示的临床试验,可以发现生物等效性试验所占比重较大,专业的临床试验机构承接的临床试验较多。
数据来源:CDE药物临床试验登记与公示平台、药智数据中国临床试验数据库
仿制药一致性评价最新进展:受理号达1289个,通过308个
截止2019年7月18日,CDE承办的一致性评价受理号已达1289个,共计373个品种,涉及383家药企;其中,289目录品种受理号611个,共计133个品种。注射剂受理号349个,共计107个品种。
详细信息请见:https://news.yaozh.com/archive/26740.html
来源:药智网
2019上半年24个创新器械产品进入CMDE“绿色通道” 国产超七成
2019年上半年(H1)共有24个创新医疗器械纳入特别审批,9个获批上市;11个医疗器械纳入优先审批,2个获批上市。
自2014年3月1日《创新医疗器械特别审批程序(试行)》和2017年1月1日《医疗器械优先审批程序》正式实施,针对特定医疗器械产品开通了绿色审批通道,加速推进了创新性强、技术含量高、临床需求迫切的医疗器械上市,同时加快了高端医疗器械进口替代的步伐。

图1 我国针对特定医疗器械产品开通了绿色审批通道
一、创新医疗器械特别审批
1. 2019年H1,共24个纳入程序,9个获批上市
2019年H1,国家药监局医疗器械技术审评中心(CMDE)共公示了7批创新医疗器械特别审批申请审查结果,共有24个医疗器械获得创新医疗器械资格认定并进入特别审批程序,另有9个创新医疗器械成功获批上市。
从产品类型来看,上半年新纳入的产品中,各类型产品占比差距不大,其中最多的无源植介入、耗材、康复及中医设备共有8个,最少的体外诊断设备和试剂有2个。
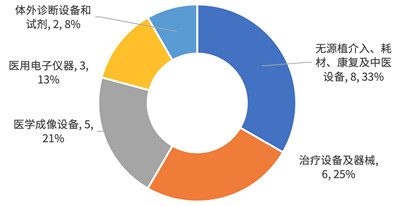
图3 2019年H1 新纳入创新特批的产品类型分布
除6个进口产品之外,国产有18个,占75%。从地域分布上,广东最多,有7个;其次为北京(4个)和上海(3个);江苏2个;浙江和陕西各拥有1个。
2. 2014—2019年H1,累计234个纳入程序,63个获批上市
累计共234个创新医疗器械产品被纳入特别审批,其中有63个获批上市。产品数量2018年增速略有下降,而从2019年上半年的情况来看,今年增速将相对稳定。见图2。
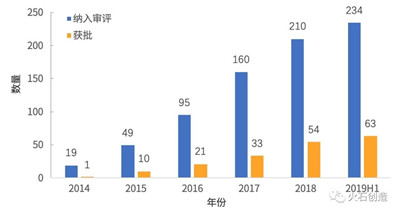
图2 2014—2019年H1创新医疗器械纳入审评与获批数量
注:获批套件类产品数量按注册证编号分开统计
从地域分布上,北京、上海、广东和江苏四省以数量上的绝对优势稳居前四,浙江排在第五,河北、天津、山东等具有一定医疗器械产业基础的省份紧随其后,可见医疗器械产业大省依然是国产医疗器械创新的排头兵。见图4。
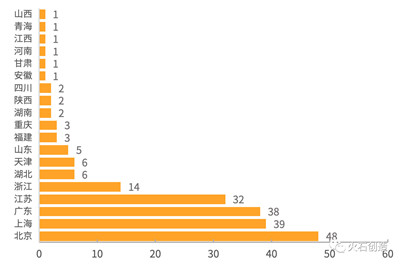
图4 2014—2019年H1创新医疗器械产品地域分布
二、医疗器械优先审批:2019年H1,共11个纳入程序,2个批准上市
2019年H1有11个医疗器械被纳入医疗器械优先审批程序,另有2个成功通过优先审批程序上市。自2017年医疗器械优先审批程序正式实施以来,已累计纳入了31个医疗器械,其中有7个产品通过该程序加速上市,见表1。
表1 自2017年已通过优先审批程序上市的医疗器械

这31个医疗器械被纳入优先评审程序的同意理由分布情况见图5,总体以临床需求和优势为导向。主要为“列入国家重点研发计划”(11个)和“临床急需且在我国尚无同品种产品获准注册的医疗器械”(10个)。
其中罗氏诊断的进口产品抗PD-L1 (SP142)兔单克隆抗体试剂(免疫组织化学法),既满足诊治恶性肿瘤且具有明显临床优势的条件,又符合临床急需且在我国尚无同品种产品获准注册的条件。

图5 2017—2019年H1医疗器械被纳入优先评审程序的同意理由分布情况
三、重磅产品介绍
1. 生物可吸收冠状动脉雷帕霉素洗脱支架系统
2019年2月,通过创新医疗器械特别审批程序上市的“生物可吸收冠状动脉雷帕霉素洗脱支架系统”(NeoVas)是由乐普医疗自主研发的重磅产品。这是国内首款获准上市的生物可吸收支架,标志着我国在可吸收支架领域的研发制造能力又步上了一个新的台阶,达到了能够领先国际的水平。
该产品的基体及药物载药涂层分别由可吸收材料左旋聚乳酸(PLLA)和外消旋聚乳酸(PDLLA)制成,支架基体和涂层在体内逐步生物降解和吸收,无永久性支架存患者体内。
2. 正电子发射及X射线计算机断层成像扫描系统
2019年5月,通过创新医疗器械特别审批程序上市的“正电子发射及X射线计算机断层成像扫描系统”是我国自主原创技术打造的全数字PET,从关键材料、核心元器件到系统整机全部为我国自主研发。
3. 多孔钽骨填充材料
2019年1月,通过创新医疗器械特别审批程序上市的“多孔钽骨填充材料”,是首个可用于四肢非承重部位的腔隙性松质骨缺损填充的金属骨填充材料,打破了美国的垄断,弥补了国内在这一领域的空白,不但可减少国内对进口骨修复材料的依赖,还将凭借领先技术进军海外市场,业内预估产值超300亿元。
四、小结
从创新医疗器械特别审批及医疗器械优先审批的情况来看,我国医疗器械产业的创新生态存在区域创新能力差异较大的问题。目前,以北京、广东和长三角地区为代表的医疗器械产业大省依然是国产医疗器械产业创新的主力军,而其他地区的创新成果则较少。
医疗器械国产化替代是一个艰难的过程,加强区域之间的交流和协同,共同提升医疗器械产业的创新生态,最终带动中国医疗器械产业实力的整体提升,将是产业面临的共同课题。
来源:火石创造
国外信息
2009-2018年六大监管机构的新药批准情况
监管环境的明显改善及跨国公司申报策略的变化使获得上市许可的所需的时间普遍减少,六大监管机构之间的协调统一性也有所提升,过去10年(2009-2018年)里获批上市的新药数量也有所增长,六大监管机构包括欧洲药品管理局(EMA)、美国食品药品监督管理局(FDA)、日本药品和医疗器械管理局(PMDA)、加拿大卫生部、瑞士医药管理局(Swissmedic)和澳大利亚药品管理局(TGA)。
国际法规科学创新研究中心(CIRS)在题为《2009-2018年6大监管机构批准的新药:关注加速审评通道和孤儿药状态》的研发简报70中发布了一项新研究,该研究对新活性物质(NAS)的获批情况进行统计,有如下发现。
六大监管机构包括欧洲药品管理局(EMA)、美国食品药品监督管理局(FDA)、日本药品和医疗器械管理局(PMDA)、加拿大卫生部、瑞士医药管理局(Swissmedic)和澳大利亚药品管理局(TGA)(如图1所示)。
CIRS的统计表明,六家监管机构批准的品种数从2009-2013年间的16个NAS增长至2014-2018年间的52个NAS,这表明在此阶段内,有更多的产品实现了国际化。
影响新药提交申请并获得监管机构批准总时间的潜在影响因素包括公司策略、审评程序的实施和类型、产品类型及其治疗领域。特别地,加速审评通道(FRP)、孤儿药指定和申请人规模是影响提交和批准策略的主要因素。
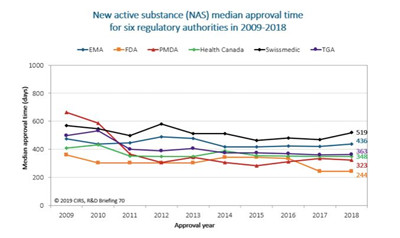
图1:监管环境的改善有助于获得上市许可所需时间的普遍缩短。(批准时间为提交申请至获得监管机构批准所需的时间。时间包括监管机构和申请人耗费的时间。EMA批准时间包括EU委员会耗费的时间。)
来源:国际法规科学创新研究中心
关注加速审评通道
六家监管机构均已实施一种特定的FRP,即旨在加速有前景新活性物质审评进程的加速审评程序(指EMA的“加速审评”、Swissmedic的“绿色通道”和另外四家监管机构的“优先审评”)。TGA自2017年开始实施优先审评体系,2018年第一次通过该体系批准新药。一般而言,通过加速审评程序所需的时间短于标准程序。
2018年,FDA(73%)加速审评所占的比例最高,其次为加拿大卫生部(35%)、PMDA(28%)、Swissmedic(13%)、EMA和TGA(10%)。
六家监管机构中,FDA提供(或可用)的加速审评通道(FRP)最多,对于有未满足医学需求的领域,加快药品的审评和/或批准,提高药品的可及性。2018年,FDA批准的NAS中有75%的品种至少从一种可用的FRP中(不包括孤儿药)获益。其他机构中,FRP的获益率在TGA的10%至加拿大卫生部的41%之间。(见图2)。

图2:六国监管机构中,FDA提供(或可用)的加速审评通道(FRP)最多,对于有未满足医学需求的领域,加快药品的审评和/或批准,提高药品的可及性。
来源:国际法规科学创新研究中心
关注孤儿药状态
六家监管机构指定为孤儿药的NAS数量从2009-2013年的25%提高至2014-2018年的38%。2018年,FDA批准的孤儿药最多(35),而PMDA的最少(8)。加拿大卫生部现在尚未执行孤儿药政策;但是,该机构2018年批准了15个FDA、EMA或TGA指定为孤儿药的NAS。
2018年,FDA的孤儿药中位批准时间最短(243日),因为其中88%的品种均通过加速审评获批。六家监管机构中,只有EMA的孤儿药中位批准时间超过非孤儿药。2018年Swissmedic的这些时间指标接近。2018年TGA批准的孤儿药中,20%通过刚启动的优先审评程序获批。
2014-2018年获得六家监管机构批准的52个NAS品种中,只有10个NAS品种在所有监管机构中被指定为孤儿药,表明各监管机构间指定孤儿药的标准有一定差异。一般而言,孤儿药NAS的中位提交时间差长于非孤儿药NAS。
事实上,大部分孤儿药NAS都是由非顶尖企业注册的,突出小规模企业在推动创新方面起到的重要作用。
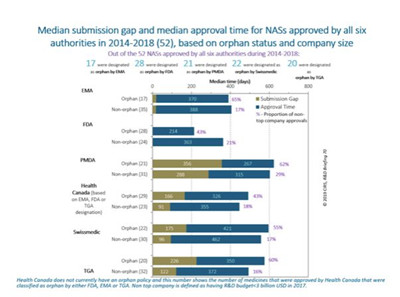
图3:CIRS的统计表明,大部分孤儿药NAS都是由非顶尖企业注册的,突出小规模企业在推动创新方面起到的重要作用。
来源:国际法规科学创新研究中心
来源:新浪医药新闻
FDA发布《辅料数据库使用指南》草案
美国 FDA 于 7 月 10 日发布《辅料数据库使用指南》草案,解释了即将对 FDA 的辅料数据库(IID)开展的一些改进,包括将增加每种辅料的最大每日暴露量(maximum daily exposure,MDE)以及对数据库更频繁地更新。指南还介绍了如何使用 IID 及其局限性。
指南表示,“将根据 GDUFA II 承诺函在未来对 IID 进行修改。到 2020 年 10 月 1 日,FDA 将完成对辅料数据库的改善提升,以便使用者可以进行电子查询,获得每种给药途径的最大每日摄入量(maximum daily intake,MDI)和最大每日暴露量信息。”多年来申办人最关心的问题与最大含量和 MDE 之间的差异相关。最大含量是一个剂量单位中的辅料量,而 MDE 则基于药品标签中建议的给药剂量计算出来。申办人从未有过确定 MDE 的好方法,因为 IID 中没有与实际产品的链接。FDA 在 GDUFA II 下的承诺之一是在 2020 年 10 月 1 日之前更新 IID 以包括 MDE。这将是 IID 自建立以来的最大改进。
美国普享药协会(AAM,原仿制药协会)曾抱怨表示,在目前的数据库形式中,最大含量一栏是没有意义的,因为最大含量的水平被设定为一种给药途径一种剂型的已获批的最大量,这些含量水平不能转化为更有用的限制,例如,其它剂型的 MDE。业内人士表示,了解 MDE 是药品配料的关键,了解已经在已获批药品中用于特定给药途径的辅料的 MDE 是有帮助的。IID 的改进将允许公司以电子方式查询获取该信息。
此外,FDA 还将每季度发布对 IID 所做更改的更新。更新通知将包括所做的每项更改,以及对于每项更改所替换的信息。这回应了过去仿制药行业对于 IID 需要更频繁更新的抱怨。
IID 最初于 1987 年以纸质形式提供,2003 年开始 FDA 以在线数据库的形式提供信息。FDA 自数据库建立以来一直在使用 IID,并承诺全年更新,提供更多最新和有用的信息。IID 中的辅料被不断添加或删除(如果存在相关安全性问题),并且在有新信息可用或新产品获批时修改相应的辅料水平。
FDA 于 2011 年成立 IID 工作组,识别 IID 的缺点并加以改进。FDA 还在 2015 年发布了一份通知,收集利益攸关者的意见,并根据这些意见发布了该指南。指南还提供了有关申请人如何优化 IID 使用的建议。
FDA 辅料数据库包括有关每种辅料的以下信息:成分名称、给药途径、剂型、美国化学文摘社(CAS)号、独特成分标识符和最大含量。工业界可能会使用 IID 中的信息来支持辅料的安全性;被列入 IID 中意味着该辅料先前已用于 FDA 批准的药品中。如果一个辅料已在获批药品中用于特定给药途径,则该辅料通常不被认为是新辅料,并且可以在下次以相同的给药途径用于新产品中时进行较少的评估。
指南还提供了术语定义,IID 是如何工作的,关于如何搜索数据的建议,并讨论了缺失值的重要性和 IID 的效用。对于使用 IID 可能产生的不同文件,指南中还列出了一个有用的联系人清单。关于如何使用数据库 , 指南指出,在论证一个辅料的使用参考 IID 时,申办人可能需要提交更多信息。“当对于一个具有多个等级的辅料参考 IID 时 , 最好指定拟议等级并参考该等级的 IID 列表,或者 , 如果无法这样做的话,需要解释参考的 IID 清单等级与拟议辅料等级之间的关联。”
来源:GMP办公室
2018年FDA数据完整性相关警告信分析
An Analysis Of 2018 FDA Warning Letters Citing Data Integrity Failures
2018年FDA数据完整性失败相关警告信分析
This article represents the fourth year I have published an evaluation of FDA warning letters associated with data governance and data integrity deficiencies. (Here are links to the 2015, 2016, and 2017 installments.) The agency’s enforcement for failures in data integrity and data governance began almost 20 years ago. This year, however, we may have turned the corner, which I will address below. Although the FDA is not the only health authority that identifies these issues in inspections and enforcement actions, their transparency ensures the data is readily available.
这篇文章呈现了我第四年发表的关于数据管理和数据完整性缺陷相关的FDA警告信的评估。FDA当局强制执行数据完整性和数据管理失败几乎开始于20年前。然而,今年我们可能到了一个拐点,我将在下面讨论。但是,FDA不是唯一在检查和执法行动中识别这些问题的卫生机构,但它们的透明度确保了数据随时可用。
In this summary, this article will identify:
本文将确定的摘要如下:
﹒Warning letters from the 2018 calendar year (CY2018) that cite data integrity deficiencies
2018年(CY2018)引用数据完整性缺陷的警告信
﹒The number of warning letters citing this topic in the past 11 years and the countries where the impacted sites are located
过去11年中引用此主题的警告信函数量以及受影响工厂所在的国家/地区
﹒The regulations identified most frequently in CY2018 drug GMP warning letters citing data integrity failures.
该法规识别出了2018年药物GMP警告信中引用数据完整性最常出现的失败。
As in past years, all data integrity deficiencies identified in Form 483s and warning letters are failures to follow cGMPs as specified in the predicate rules. The FDA has not implemented novel interpretations or requirements applicable to data governance. The use of computer systems and other electronic systems requires different approaches to ensure compliant practices, but these are all based on the existing regulations in 21 CFR 211.
与过去几年一样,483表和警告信中确定的所有数据完整性缺陷都是未遵循法规规定的cGMP。FDA尚未实施适用于数据管理的新颖解释或要求。使用计算机系统和其他电子系统需要不同的方法来确保合规实践,但这些都是基于21 CFR 211中现有的规定。
Data Integrity GMP Warning Letters And Trends From The Past 11 Years
数据完整性GMP警告信和过去11年的趋势
Table 1 identifies the FDA warning letters issued to drug manufacturers in CY2018 that include data integrity deficiencies. The table includes the date of issuance and the country where the cited facility is located. The FDA issued 85 drug GMP warning letters in CY2018, excluding those issued to compounding pharmacies and outsourcing facilities. Forty-two of the 85 included a data integrity component, for a total of 49 percent of the warning letters. No warning letters were posted in December due to the partial government shutdown.
表1列出了在2018年向药品生产商发出的包含数据完整性缺陷的FDA警告信。该表包含了发布日期和所引用工厂所在的国家。美国FDA 2018年发布了85份药品GMP警告信,不包括发给综合药房和外包工厂的警告信。85个中的42个包含数据完整性内容,占总警告信的49%。由于政府部分关闭,12月没有发布警告信。
Table 1: CY2018 Drug Warning Letters With Data Integrity Deficiencies
表1:2018年包含数据完整性缺陷的FDA警告信

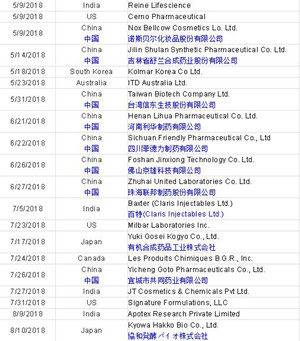
Table 2 and Figure 2 present the number of data integrity-associated warning letters by country over the last 11 years, CY2008 through CY2018, along with a cumulative total. The number of warning letters referencing this topic ranged from four to six from 2008 through 2013 and doubled in CY2014 to 10. The number of warning letters increased from 15 in 2015 to 41 in 2016, and then to 56 in 2017. In 2018, the number actually decreased 25 percent, to 42.
表2和图2显示了过去11年(2008至2018)各国与数据完整性相关的警告信数量以及累计总数。从2008年到2013年,关于该主题的警告信函数量从4个到6个不等,而在2014年翻了一番达到10个。警告信的数量从2015年的15个增加到2016年的41个,然后在2017年增加到56个。2018年,数字实际上减少了25%,达到42。
The number of countries associated with these warning letters continues to increase. In 2018, the sites that were the subject of warning letters were in 11 different countries. Figure 2 also shows that nearly 80 percent of data integrity-related warning letters issued since 2008 occurred in the past four calendar years. The number peaked in CY2017, and it will be interesting to see if the number decreases again in CY2019, as it did in CY2018.
与这些警告信相关的国家数量继续增加。在2018年,作为警告信主体分布在11个不同的国家。图2还显示,自2008年以来发布的近80%与数据完整性相关的警告信发生在过去四年。这个数字在CY2017达到顶峰,有趣的是看看这个数字是否会在CY2019中再次下降,就像在CY2018中那样。
Figure 3 shows the data integrity-associated warning letters by country from CY2015 through CY2018. South Korea is new to this group in the past two years. Canada and Mexico have been members since 2010 and 2012 respectively. Singapore joined in 2017, and Australia, Taiwan, and the Dominican Republic were new to the group in 2018.
图3显示了从2015年到2015年CY2018各国的数据完整性相关警告信。韩国是过去的两年新加入的。加拿大和墨西哥分别从2010年和2012年开始成为其中的一员。新加坡于2017年加入,澳大利亚,台湾和多米尼加共和国于2018年成为其中的新成员。
Table 2: Number of Data Integrity Associated Warning Letters by Country, CY2008–CY2018
表2:按国家划分的数据完整性相关警告信的数量,2008-2018
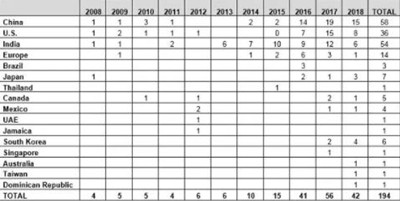


Table 3 compares the number and percentage of warning letters citing data governance and data integrity in the past 11 years compared to the most recent four years. China tops the list in both the last four years and the last 11 years. In the past four years, China significantly outperforms India in this area, and the U.S. comes in third. Europe remains constant at approximately 8 percent of the total for both periods, and the rest of the world (ROW) is constant at approximately 16 percent of the totals.
表3比较了过去11年与最近四年,引用数据管理和数据完整性的警告信函的数量和百分比。中国分别在过去四年和过去11年中都名列榜首。在过去的四年中,中国在这一领域明显高于印度,而美国排名第三。欧洲在两个时期内均保持占总数的8%左右,而世界其他地区(ROW)则持续约占总数的16%。
Table 3: Geographic Totals and Percentage, 2015–2018 and 2008–2018
表3:2015 - 2018年和2008 - 2018年的地理上的总量和百分比
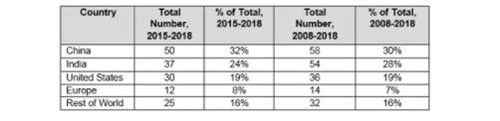
Table 4 shows the regulations most frequently cited in the warning letters in CY2018. Many of the deficiencies did not identify a regulation or were provided by the FDA as “conclusions” or “data integrity remediation” instructions to which the firms must respond. Warning letters issued to API manufacturers do not identify 21 CFR 211. The citation of regulations continues to follow the FDA’s stated goal of focusing on the evaluation of predicate rule requirements.
表4列出了2018年警告信中最常引用的法规。许多缺陷没有明确法规条款,或仅是FDA在“结论”或“数据完整性纠正”中引用的要求公司必须回复的说明。发给API制造商的警告信则没有指明21 CFR 211。法规的引用仍然遵循FDA关于评估规则要求的既定目标。
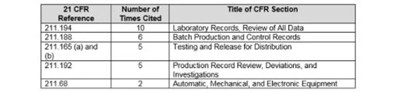
Table 4: Regulations Most Frequently Cited in CY2018 Data Integrity-Associated Drug Warning Letters
表4:2018年数据完整性相关的药物警告信中最常被引用的法规
Actions Firms Can Take To Prevent, Identify, And Remediate Issues
公司预防,识别和纠正问题可以采取的措施
The number of data integrity-associated warning letters decreased significantly between CY2017 and CY2018, though the percentage in 2018 remains slightly above that for CY2016. We will follow the trends for CY2019 to see if the number of data integrity-related warning letters continues to decrease.
数据完整性相关警告信函的数量在2017和2018年之间显着下降,但2018年的百分比仍然略高于2016年。我们将跟踪2019年的趋势,看与数据完整性相关的警告信的数量是否继续减少。
So, how should firms prevent, detect, and remediate these problems before the FDA or other health authorities become involved? My advice remains virtually unchanged from last year. My recommendations are divided into those for executive management and those for functional areas. A focus on management of contract services is included among the actions for firms to consider. Additional detail on contract manufacture and data governance is provided in two previous articles published in 2017.
那么,在FDA或其他卫生部门介入之前,企业应该如何预防,检测和纠正这些问题呢?我的建议与去年相比几乎没有变化。我的建议分为高级管理层和职能区域。在这些要考虑的行动中,公司需要关注外包服务管理。有关外包生产和数据管理的更多详细信息,请参见2017年发布的两篇文章。
Executive Management Ownership
高级管理层所有权
﹒Executive management must develop and reinforce a culture of quality.
高级管理层必须发展和加强质量文化
﹒Executive management must establish and maintain a corporate culture of openness where employees may report problems and failures without fear of retribution. In fact, reporting of problems should be encouraged and rewarded.
高级管理层必须建立并维护一种开放的企业文化,员工可以不必担心报复地报告问题和失败。事实上,报告问题应该被鼓励和奖励。
﹒Executive management must own the gap assessment process and remediation efforts. Remediation may be costly and time-consuming. Firms often uncover additional problems along the way. Don’t expect to complete remediation quickly; it’s often a multiyear process.
高级管理层必须拥有差距评估流程和纠正措施。纠正的过程可能是昂贵且耗时的。公司经常会在中途发现其他问题。不要指望快速完成补救; 这通常是一个多年的过程。
Technical Area Actions
技术领域行动
﹒Cross-functional teams should perform gap assessments for both paper and computer systems against predicate rule requirements and specific data governance/integrity guidance from health authorities. The team should identify corrective actions and a timeline for their implementation. Firms should implement interim corrective actions until they can put fully compliant solutions in place.
一个跨职能团队根据既定法规要求和卫生当局的特定数据管理/完整性指南,对纸质和计算机系统进行差距分析。团队应确定纠正措施和实施时间表。在实施完全合规的解决方案前,企业应该实施临时纠正措施。
﹒Firms should map data and process flows and identify and remediate areas of risk. Results from this exercise can contribute to the gap assessments described above.
企业应该绘制数据和工艺流程图,并识别和纠正风险。该做法的结果可有助于上述差距分析。
﹒Firms should validate systems for their intended purpose and ensure that adequate controls are in place to ensure that deleted or altered data can be detected. Purchasing software that the supplier claims is Part 11 compliant does not suffice.
公司应对系统进行验证以符合其既定用途,并确保采取适当的控制措施,以确保删除或更改数据的行为可以被检测到。采购的软件仅仅供应商声称符合Part 11是不够的。
﹒Monitor enforcement actions including Form 483s, warning letters, import alerts, EU reports of GMP noncompliance, and World Health Organization (WHO) Notices of Concern. All of these, except for the Form 483s, are available without cost on the internet, and Form 483s are available from several commercial sources. A selected subset may be found on the FDA website.
执法行动跟踪,包括483表,警告信,进口警报,欧盟GMP不合规报告和世界卫生组织(WHO)关注事项通知。除了483表之外,所有这些都可以在互联网上免费获得,483表可以从一些商业渠道获得。可以在FDA网站上找到经挑选的部分。
﹒Ensure that the data governance processes at suppliers and contract service providers are adequate to ensure that data is valid and trustworthy. This effort begins with rigorous due diligence evaluations, periodic on-site oversight, and appropriately detailed quality agreements. Contractors must have procedures and processes to ensure integrity of the data they produce.
确保供应商和合同服务提供商的数据管理流程足以确保数据有效且可信。这项工作始于严格的尽职调查评估,定期现场监督以及恰当详细的质量协议。承包商必须拥有以确保其生成的数据的完整性的程序和流程。
Conclusion
结论
Data integrity and data governance remain as enforcement initiatives of global health authorities. The U.K.’s Medicines and Healthcare Products Regulatory Agency (MHRA) was the earliest to enter the area with its 2015 guidance and 2018 published revision. The European Medicines Agency (EMA), WHO, Pharmaceutical Inspection Co-operation Scheme (PIC/S), Australia, Canada, and China followed in 2016. Further, enforcement is not limited to the GMP area but includes good clinical practice (GCP), with the most impactful cases at sites that perform bioavailability and bioequivalence studies. For these firms, the data for hundreds of products is impacted. Sponsors must frequently repeat the studies at different sites. Among the more significant failures in this area were identified at GVK and Semler Research. Consequences at Semler included a three-page Form 483, untitled letter, WHO notice of concern, and EMA recommendation of suspension.
数据完整性和数据管理仍然是全球卫生当局的执法行动。英国的药品和保健品监管机构(MHRA)是最早进入该领域的,出版了2015年指指南和2018年修订版。欧洲药品管理局(EMA),世界卫生组织,药品检查合作计划(PIC / S),澳大利亚,加拿大和中国于2016年紧随其后。此外,执法不仅限于GMP领域,还包括良好临床规范(GCP),在进行生物利用度和生物等效性研究方面是最具影响力的案例。对于这些公司,数百种产品的数据会受到影响。申报方必须经常在不同地点重复研究。在GVK和Semler Research中发现了该领域中更多重大的失败。Semler的结果包括一份三页的483表,无标题信,世卫组织关注事项通知以及EMA的暂停建议。
GMP enforcement citing data governance and data integrity is still significant, expanding in its geographic distribution. Deficiencies in data governance and data integrity have remained markedly consistent over the 11 years addressed in this report, with a few new areas identified each year. Newer focus areas that appeared in 2017 continued in 2018 and include:
GMP实施引用数据管理和数据完整性仍然很重要,其分布的地理范围也在不断扩大。本报告所述的数据管理和数据完整性方面的缺陷,在11年中始终保持显着一致,每年都会确定一些新的领域。在2017年出现且2018年继续的较新焦点领域包括:
﹒Firms that repackage APIs were transferring analytical results onto certificates of analysis on their own letterhead, making it appear that they generated the results. The practice obscures the supply chain from the company that purchases and uses the material in the manufacture of drug products.
重新包装API的公司将检验结果转移到有他们自己信头的检验报告上,使他们看起来产生了结果。这种做法向购买和使用药品生产所用的材料的公司掩盖了供应链。
﹒Firms aborted an excessive number of analytical of runs.
企业中止了很多的分析测试。
﹒Firms manipulated “integration suppression” parameters within chromatography data systems, intending to obscure or minimize impurity peaks.
企业在色谱数据系统中操纵“积分抑制”参数,旨在模糊或最小化杂质峰。
I expect this type of problem to expand in scope to more OTC manufacturers because action in this area is a clear trend that began in 2017. I will also watch for this topic to be cited more frequently in enforcement actions taken against compounding pharmacies and outsourcing facilities. Previously, most of the problems in this area addressed failures in aseptic processing, including facilities and equipment issues. I look for data integrity to be cited more frequently in both Form 483s and warning letters issued to these firms.
我预计这类问题会扩大到更多OTC制造商的范围,因为从2017年开始,这一领域的行动趋势就已明确。我还将关注在针对综合药房和外包设施的执法行动中,这一主题被更频繁地引用。此前,该领域的大多数问题都是无菌过程的失败,包括设施和设备问题。我发现在发给这些公司的483表和警告信中更频繁地引用了数据完整性。
来源:GMP办公室
日本医疗器械监管概览
2014年,日本医疗器械市场年销售额约325亿美元,随着日本人口老龄化情况日益严重,2018年日本医疗器械市场销售额已激增至478亿美元,增长势头惊人。据统计,日本的医疗器械产品中59%为国内自产,41%从海外进口。
审评审批体系
日本医疗器械产品分类采用国际通用的四级分类法,即高度管理医疗器械(Ⅲ类、Ⅳ类)、管理医疗器械(Ⅱ类)、一般医疗器械( Ⅰ类)。一般医疗器械由备案人向审评机构PMDA申请备案;管理医疗器械是在认证基准确定的情况下,由第三方认证机构进行认证,对于没有认证基准的或者不符合认证基准的管理医疗器械,由PMDA进行审评,厚生劳动省(以下简称厚生省)承认;高度管理医疗器械由PMDA进行审评,厚生省承认。但对于有承认基准和审查指导原则的高度管理医疗器械,也可由第三方认证机构进行认证。申请认证的产品如不符合承认基准或审查指导原则,需要向厚生省申请承认。
第三方机构必须符合《医疗器械 质量管理体系 用于法规的要求》(ISO13485)中的相关规定,并对医疗器械产业非常熟悉,对加工场所、产品质量等具有丰富经验的专业公司才有资格成为第三方审核机构。产品核准上市后,第三方机构还须负责该产品的事后监督,即质量标准执行情况的检查等工作,而绝不是“一审了之”。厚生省负责对第三方机构资质以及产品审核情况等进行监督管理。一旦发现第三方机构在审核中存在违规操作情况,厚生省有权取消其认证医疗器械产品的资格,并视情节严重程度予以经济处罚。
产品评估体系
日本医疗器械产品评估体系与美国的医疗器械产品评估体系相似。例如,厚生省在签批上市许可证之前,首先要对该产品的质量、有效性、安全性等进行综合性评估。其次,主管医疗器械产品审批的政府机构会派人到申请产品上市的生产企业实地了解生产线情况及生产商的资质情况。对于新批准上市的医疗器械产品,监管部门会定期抽查产品进行复核,尤其是拥有新设计、新原理、新结构的创新医疗器械产品,监管部门会在批准其上市4年后再次抽查其产品,并检查其质量、有效性与安全性是否与当初申报时相同;新用途医疗器械产品一般在3年后进行复查。
上市后监管体系
在参考欧美国家对医疗器械产品上市后监管体系法律法规的基础上,日本政府制定了一套符合本国国情的医疗器械上市后监管体系,最重要的一点是:对于每件获准上市的器械产品,生产商必须保证其质量100%合格,且安全有效;一旦产品上市后被发现存在重大质量问题或安全隐患则由厂商自己负责整改。鉴于此,日本新版医疗器械上市后监管法特别规定:生产商必须建立本公司的质量控制小组和产品安全负责人制度,一旦产品投放市场后出现问题,则这两个小组的负责人将被首先问责。
据了解,日本从2015年开始实施新版《药品医疗器械产品及再生/细胞疗法产品质量安全法》。过去在日本上市的所有医疗器械产品均需持有详细的QMS(质量管理体系)文件,而新版法规则对此作了简化处理——生产商只要有QMS证书并提供产品确实是按照QMS生产的说明即可。但厚生省会对医疗器械厂商进行不定期的飞行检查。
来源:中国医药报